Advances in Animal and Veterinary Sciences
Mini–Review Article
Advances in Animal and Veterinary Sciences. 2 (4S): 1 – 10Special Issue – 4 (2014) (Reviews on Frontiers in Animal and Veterinary Sciences)
Basic Techniques and Limitations in Establishing Cell Culture: a Mini Review
Priyabrat Swain1*, Pramod Kumar Nanda2, Sukanta Kumar Nayak1, Sudhansu Sekhar Mishra1
- Central Institute of Freshwater Aquaculture, Kausalyaganga, Bhubaneswar–751 002, India
- Indian Veterinary Research Institute, Eastern Regional Station, Belgachia, Kolkata–700 037, India
*Corresponding author:pswainy2k@yahoo.co.in
ARTICLE CITATION:
Swain P, Nanda PK, Nayak SK, Mishra SS (2014). Basic techniques and limitations in establishing cell culture: a mini review. Adv. Anim. Vet. Sci. 2 (4S): 1 – 10.
Received: 2014–03–07, Revised: 2014–03–26, Accepted: 2014–03–27
The electronic version of this article is the complete one and can be found online at
(
http://dx.doi.org/10.14737/journal.aavs/2014/2.4s.1.10
)
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
ABSTRACT
Cell culture has become an indispensable tool in modern day science and is being innumerable applied in both fundamental and applied biological research. This technology involves isolation of cells from their natural (in vivo) location and propagation in vitro in favourable artificial conditions. Cell culture can either be short term (primary culture) or long term depending upon purposes. In several cases, cells are grown for short terms i.e., for few days or weeks to obtain cells from target organs or tissues to pursue specific purposes. On the contrary, the long–term culture of cells is done to establish cell lines. Irrespective of the purpose and culture practices; optimal culture conditions in form of nutrients, media, supplements and growth factors is required to be maintained under controlled environment for in vitro growth of cells. Over the years, appropriate methods have been developed and standardized for culture of cells from all most all the tissues and organs of diversified species. However, the basic principles of cell culture techniques remain mostly unchanged since its introduction. This mini review illustrates the aspects that are critically required for cell culture and also focuses on the limitations, especially adaptation of cells to artificial environment for optimal growth and differentiation in order to form an established cell culture.
Cell culture is the complex process which involves isolation or removal of cells from their natural (in vivo) and their subsequent growth under controlled conditions in an artificial (in vitro) environment. Cultured cells, either as short term or as established cell lines, from a broad range of tissues and organs have many applications in fundamental and applied biological research (Keay, 1975; Hightower and Renfro, 1998; Marrec–Croq et al., 1999; Butler, 2005; Nema and Khare, 2012). These in vitro models form the biological basis of most alternative methods (Hartung, 2007) and are used to study cell differentiation, toxicology and pharmacology, cancer research thereby avoiding the use of laboratory animals. Further, cell culture is widely used as a diagnostic and research tool, particularly for diagnosis of viral diseases (Wolff, 1988) as isolation of pathogenic viruses from animals are totally dependent on the availability of a live host such as permissive cell cultures (Hetrick and Hedrick, 1993). Cell cultures are also used in vaccine manufacturing using recombinant DNA (rDNA) technology and other biological products such as production of immunobiologicals (monoclonal antibody, recombinant protein, lymphokines), cell and gene therapy, novel drugs selection and improvement etc. (Butler, 2005; Eibl et al., 2009; Nema and Khare, 2012). Because of such broad range of applications, there is a growing need to develop and establish short term and continuous cultures from various commercially important animal species. The in vitro growth of cells requires the essential features of the conditions within the body that cells experience in living tissues for their optimal cell survival, growth and function (Cooke et al., 2008). To achieve this, optimisation of the in vitro culture conditions providing culture media, substrate, nutrients and growth supplements is required making a conducive environment for cells to function, grow and propagate in a realistic manner. This mini review emphasizes the role of various factors and also focuses on the limitations in establishing cell culture.
Tissue or cell culture as a technique was first devised at the beginning of the 20th century as a method to study the behaviour of animal cells in vitro. Prior to this, Roux (1885), an embryologist, was able to maintain embryonic chicken cells in warm saline for several days and established the principles of tissue culture. Carrel and Burrows (1911) demonstrated that the survival of cells isolated from blood and connective tissue can be made in serum and plasma. Harrison (1907) established the methodology of tissue culture and observed cell growth in clotted lymph fluid from explants of frog embryo tissue by the “hanging drop” method. This was followed by the work of Carrel and his co–worker who succeeded in growing adult and embryonic tissues of warm blooded mammalian cells in vitro (Carrel and Burrows, 1910). Carrel (1912) expanded the possibilities of cell culture by keeping fragments of chicken embryo heart alive for over 3 months in vitro. To overcome the contamination problems, Carrel or T–flask, developed by Carrel and his co–worker in 1923, has become one of the important items of cell culture laboratories enabling the subculture of cells (Butler, 1991). The technique of trypsinization was exploited by Rous and Jones (1916) using trypsin (a proteolytic enzyme) to dissociate cells from tissue matrix. This facilitated the subculture of adherent cells to produce homogeneous cell strains and marked the start of animal cell culture in true sense (Moscona, 1952). Till to that period, culture of cells from primary explants of tissue dominated the field (Fisher, 1925). The developments that occurred in over ten years (1940–1950) made cell culture widely available and accepted as a tool for scientists. Notable among these are use of antibiotics to avoid contamination problems in cell culture and development of chemically defined culture media by Eagle (1955). These advancements in cell culture technology offered more scope to researchers to widely use cell, tissue and organ culture in their research as a result of which cell culture is routinely carried out in many laboratories throughout the world.
Cell culture models are highly defined system, in which parameters and conditions can be regulated leading to more reproducible results in comparison to other studies. Over the last decades, these models are extensively used to study morphological, biochemical signaling processes, toxicity testing, study of reconstructed tissue equivalents with aim to reproduce the in vivo environment (Carere et al., 2002), transfection of cell lines with specific enzymes to study the biotransformation of compounds (Yoshitomi et al., 2001; Hashizume et al., 2009). Some of the types of cell culture models and or their applications are as follows.
Different in vitro cell culture models can provide an insight into the mechanisms of infection for various microbial pathogens (Duell et al., 2011). Of late, several culture models are available for studying neurodegenerative diseases in order to gain a better understanding of the disease and understand their underlying mechanisms to investigate new therapeutic strategies (Figlewiczet al., 2000). Further, these in vitro cell culture models might provide important insights about the pathogenesis of many disorders viz. cervical dysplasia–pluripotent stem cell models of human heart disease, human ES cell lines for Alzheimer’s disease drug discovery, cell culture model for Huntington’s disease and may also replace in vivo models to evaluate compounds or the screening of potential pharmacological agents against many disease conditions. Currently, a fast, efficient and reproducible cultured cell system from animal models for the screening and testing of compounds for the treatment and therapy of Abeta–associated diseases (AD) has been developed (Trinchese et al., 2004). Furthermore, cell culture models are reported to be versatile and extremely useful experimental models to investigate the biology of both the normal and the pathological isoform of the prion protein of transmissible spongiform encephalopathies /prion diseases. However, only few cell culture models permissive to prion replication are available till date. Among them, mouse neuroblastoma cell lines (N2a) are most commonly used for screening as well as selecting possible therapeutic compounds so as to develop new strategies for the investigating specific diagnostic markers (Lawson et al., 2008).
Cell culture models have several advantages over animal model for studying microbial pathogenesis and are now being considered as indispensable tools to study bacteria–induced cell death such as apoptosis or necrosis, lysis, adhesion, invasion, and cell–to–cell movement etc., of different foodborne pathogens like Listeria monocytogenes, E. coli, Clostridium perfringens, Vibrio sp., Shigella sp., etc. Further, cytopathic effects and multiplication of avian flu virus (H5N1) and hepatitis A virus can also be studied using cell culture models (Salyers and Whitt, 2002; Bhunia and Wampler, 2005).
Cell culture model is mostly used for oxidative stress related studies (Gille and Joenje, 1992), to test therapies against hyperglycemia–mediated oxidative stress and injury (Vincent et al., 2005), mitochondrial dysfunction of familial amyotrophic lateral sclerosis (Menzies et al., 2002) in cellular studies.
Such types of models are successfully used in studying endothelial barrier permeability studies, ocular barriers studies, drug absorption and delivery (Garcia et al., 2011; Schneider et al., 2013).
Microfluidic techniques facilitate simultaneous manipulation and analysis of cultured cells, starting from a single cell to larger populations, intact tissues and used for drug research (Wu et al., 2010). With the recent advances, this technology has several advantages over conventional cell culture methods and holds great promise for the creation of advanced cell culture models (Meyvantsson and Beebe, 2008; Inamdar and Borenstein, 2011).
Cell culture models are also used to understand the mechanisms of cellular senescence and their impact on human ageing process (de Magalhaes, 2004). In this aspect, the most widely used cell culture models for studying ageing is Hayflick's.
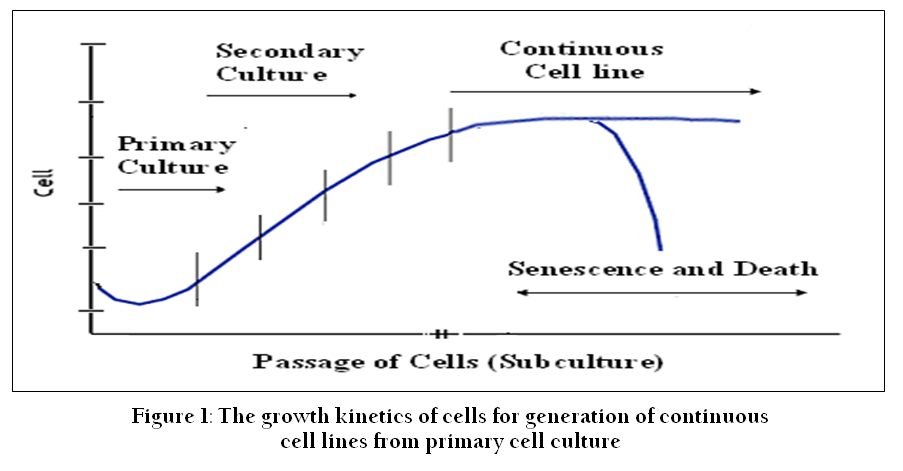
Figure 1: The growth kinetics of cells for generation of continuous cell lines from primary cell culture
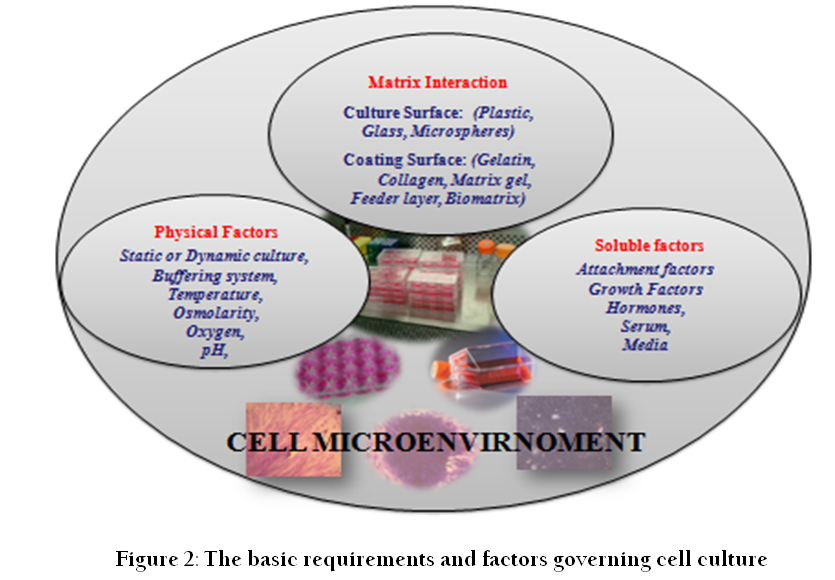
Cell culture refers to the removal of cells from an animal or plant and their propagation and cultivation in vitro in a suitable artificial environment. The process starts with primary culture of cells to achieve confluence or formation of monolayer in a culture flask supplemented with nutrients and growth factors. After achieving confluence, cells are subcultured or passaged routinely from primary to secondary and secondary to tertiary till establishment of a continuous cell line (Figure 1). During this process of culture and subcultures, several factors regulate the success of cell culture practices. To establish primary cultures or continuous cell lines, culture conditions are required to be maintained in culture flask supplemented with all the essential nutrients, supplements, hormones and growth factors that are required for metabolic activities, growth and proliferation of cells (Bettger and McKeehan, 1986; Butler and Jenkins, 1989; Davis, 2002). This, in turn, provides the microenvironment for in vitro growth of cells mimicking in vivo physiological conditions and maintaining the physical and chemical factors such as buffering systems, pH, oxygen, osmolality etc. (Figure 2).
The initial stage to consider in cell culture is the isolation of the appropriate tissue. In general, cultures from embryonic tissues survive and proliferate in a better way as compared to those from the adult (Burgess et al., 1993), as they often have a lower growth fraction and a high proportion of non–replicating specialized cells due to the age and developmental stage of the animal. Furthermore, initiation and propagation of culture is difficult, and in most of the cases, the lifespan of the culture is often shorter. From this, it is clear that more dividing cells can be obtained from gonads, embryos than from skeletal muscle, and adult animals (Nicholson, 1989; Crane, 1999). But the choice of tissue and establishment of primary culture or cell line species depends upon the purpose of its application in cellular and molecular biology.
Cell cultures may be initiated by aseptic collection of normal, embryonic, malignant tissue, or from the organ of interest by surgical or enzymatic method and placing into a conducive culture environment for attachment, growth and proliferation. This is called a primary cell culture. This is the stage of culture after isolation but before the first subculture or passage of the cells (Freshney, 2010). To obtain primary cultures, explant and tissue dissociation (mechanical and enzymatic) methods are generally employed (Bols, 1991).
The culture of cells adopting explant method dominated the field of tissue culture for more than 50 years (Fisher, 1925; Parker, 1961). This is one of the simplest procedures, which involve finely chopping the tissue (explants of tissue) into fragments of no larger than 1–2 mm3. The tissue fragments (explants) are placed to a glass or treated plastic culture flask and once attached, further media is added to cover the explants completely (Wolf and Ahne, 1982). If successful, cells will migrate out from the explants and cover the growth surface eventually forming a culture. Attachment of the explants may also be promoted by treating the plastic culture flask with substrates or coating materials like collagen, fibronectin, gelatin, polylysine, other extra cellular matrix products (Mazia et al., 1975; Crouch et al., 1987) or treating with spent medium from another culture (Stampfer et al., 1980). One of the principal advantages of this method is that some aspects of the tissue's architecture can be preserved within the explant and the biochemical functions and hormonal responses of the tissue as closely as possible to those in vivo (Dils, 1994).
The most common method for cell isolation is the enzymatic digestion. In this method, suspension of cells is obtained by chopping the tissue or organs into small fragments followed by treatment with proteolytic enzymes, which may then be cultured as a monolayer on a solid substrate, or as a suspension in the culture medium (Unchern, 1999). The most obvious advantage of cell culture and of enzymatic dissociation in particular is that it makes individual living cells accessible and can be expanded to replicate cultures.
Proteolytic enzymes (also termed peptidases, proteases and proteinases) are capable of hydrolyzing peptide bonds in proteins (Mótyán et al., 2013). The purpose is that these enzymes digest connective tissue and the components of the surrounding extracellular matrix facilitating the release of cells from a wide variety of tissues. Several enzymes viz. trypsin, collagenase, dispase, protease, pronase E, elastase, hyaluronidase are available in the market for the detachment of cultured cells, cell dissociation and cell component or membrane–associated protein isolation (Unchern, 1999). In this regard, crude enzymes are more effective but more toxic due to contamination with other proteases and prolonged exposure or treatment of cells with such enzymes can weaken their cell membranes and decrease their ability to attach to the substrate (Mckeehan, 1977; Smets et al., 1979; Pleskach et al., 1994). On the other hand, increasing the purity of an enzyme gives better control and less toxicity but may result in less disaggregation activity (Freshney, 2010). Further, enzymatic dissociation using different proteases may generate differential yield, viability and efficiency of attachment of cells, as proteases differ in their specificities (Peakman et al., 1994). The efficacy of enzymes may also vary depending upon not only type of tissue, species and age but also due to the variations in time and temperature. Therefore, all these enzymes commercially available are not suitable for every tissue type. As there are variations, an optimized method must be developed for each species as well as for each tissue type for successful primary culture.
Cell isolation can also be achieved through mechanical dispersion using a pipette or syringe. By forcing the tissue through a sieve or syringe, individual or small clumps of cells can be obtained from the bulk of the tissue. Although, this technique produces cell suspension quicker than other methods, an adhesion factor is sometimes needed to assist their attachment to the substrate as the ability of cells to attach is reduced (Toullec, 1999). Further, this procedure causes a great deal of mechanical damage and produces less viable cells than any other method (Freshney, 2010). This method is recommended for soft tissues like brain and spleen.
To start with cell culture, media has an important role to play as cultured cells need either a completely natural or an artificial medium supplemented with some natural products for growth. In early days, natural media such as biological fluids (plasma, serum, lymph, amniotic fluid etc.), tissue extract (extract of chick embryo, liver, spleen, bone marrow and leucocytes) obtained from various biological sources were used to establish cell culture (Shenoy, 2007; Nema and Khare, 2012). The major disadvantage of using natural media is that it often results in poor reproducibility in cell culture due to lack in knowledge about its exact composition.
Artificial or synthetic culture media is constituted based on two parts: a basal nutrient medium and supplements. The balanced salt solutions (BSS) such as Dulbecco’s PBS, Hank’s BSS, Earle’s BSS, phosphate buffered saline (PBS) form the basis of complex media. Besides, a complete culture media contains varying amounts and concentrations of nutrients, serum proteins, carbohydrates, and buffering agents including trace elements, amino acids, and vitamins either in a powder form or in an aqueous solution (Morgan et al., 1950). This complex mixture only provides energy but also maintains physiological conditions influencing the growth of cells in vitro (Ham, 1984; Freshney, 2010).
The media most commonly used to establish cell culture are Medium 199 (M–199), Dulbecco’s Modified Eagle’s Medium (DMEM), Eagle’s Minimum Essential Medium (E–MEM), Hank’s MEM (H–MEM), Iscove’s Modified Dulbecco’s Medium (IMDM), Ham’s nutrient mixtures (F–10 and F–12), Leibovitz (L–15), Roswell Park Memorial Institute (RPMI)–1640 and Glasgow MEM, (Lester, 2007; Arora, 2013). But apart from being a nutrient source, the media in use should also be sterile, isotonic, provide buffering capacity and maintain physiological conditions of culture. To achieve the correct pH and osmolarity, each type of media has a recommended bicarbonate concentration and CO2 tensions. On the other hand, the HEPES (4–(2–hydroxyethyl)–1–piperazineethanesulfonic acid) buffered medium is suitable, when culturing cells in atmospheric air to maintain cell viability. Therefore, selecting appropriate media for a particular cell type is required during cell culture, as their characteristics and compositions vary. For example, M–199 developed by Morgan et al., (1950), is used for culturing chick embryo fibroblasts. DMEM, a modified version of Eagle’s medium (Eagle, 1955) and created by Dulbecco and Freeman (1959) is used to achieve high–density growth of cultured cells. In contrast, F–12 formulated by Ham (1965) is required for clonal growth of low–density cells whereas Roswell Park Memorial Institute (RPMI 1640) medium is recommended for lymphoblastoid cell lines. On the other hand, L–15, a medium developed by Leibovitz (1963), contains galactose and sodium pyruvate instead of glucose and the cells can be grown without CO2. As the source of culture medium can have a major impact on the health and viability of cultured cells (Allen et al., 1985; Dodson and Mathison, 1988), each laboratory must determine the basal nutrient medium that suits best to the cell type depending upon the objective of culture (growth, survival, differentiation, production of desired proteins).
Serum, a mixture of hundreds of proteins, is a traditional supplement for mammalian cell cultures (Paranjape, 2004). Serum not only acts a source of several essential factors, attachment factors, but also improves the spreading of cells on the plastic culture substrate (Holmes, 1967; Yamada and Olden, 1978) and provides nutrients and hormones stimulating their growth (Mckeehan et al., 1976). Serum modifies physico–chemical properties (viscosity and osmolality) of the culture medium and helps in protecting labile essential nutrients in it (Ham, 1981). Besides, serum also maintains pH buffer (Barnes and Sato, 1980; Cheng et al., 1993) and inhibits activity of proteolytic enzyme such as trypsin.
Among different commercially available sera, fetal bovine serum (FBS) and newborn calf serum (NBCS) are most common and widely used in media to carry out cell culture work of various species (Wolf and Quimby, 1969; Amemiya et al., 1984; Ostrander et al., 1995). But due to high demand, limited availability and high cost, culture of cells using sera of other species such as goat, horse and chicken have also been done by several workers (Chaudhuri and Chakravarty, 1983; Castillo et al., 1991; Mcfarland, 1992; Nanda et al., 2009). Although serum offers many advantages, its constituents vary from lot to lot and may contain inadequate levels of cell–specific growth factors and an over abundance of others which may be cytotoxic resulting in poor growth of cells (Cheng et al., 1993; Freshney, 2010). Further, there is a risk of contamination with infectious agents (viruses, prions, fungi and mycoplasma or degradative enzymes. To overcome the problems of cytotoxicity, contamination, non–availability and ethical issues, a wide range of serum–free, chemically defined, animal–derived component free (ADCF); protein free media are available (Hamilton and Ham, 1977; Barnes and Sato, 1980; Stoll et al., 1996; Zimmerman et al., 2000; Brunner et al., 2010) for use in cell culture. But these media, apart from being costly, require supplementation with special ingredients to function as cell nutrients (Whitford, 2005).
The importance of amino acids in culture media for survival, proliferation and growth of animal cells of cells in vitro has long been recognized as cells cannot synthesize these by themselves (Ehrensvard et al., 1949; Eagle, 1959). Out of many amino acids available, glutamine is most commonly used as additive in cell culture and it serves as a secondary energy and carbon source for metabolism (Lane and Bennett, 1987; Newsholme et al., 2003). But L–glutamine is only stable for about 3 weeks at 4 0C in cell culture medium and converts to a form that cannot be used by cells. Further, its stability depends upon time, temperature, and pH of the solution. Therefore, most commercially available media are formulated with free L–glutamine, which is added to media just before use at varying concentrations (up to 4mM) depending upon with cell culture type and duration (Pasieka and Morgan, 1959; Arora, 2013).
One of the major problems encountered during cell culture is contamination of culture from microbes and/or fungi, either during dissection and tissue preparation or subsequent culture (Vierck et al., 2000). As the fungi and bacteria grow fast, they can be evident through visualization due to a sudden increase in turbidity and change in colour of media, affect the growth kinetics, viability and cell numbers, deplete nutrients level, add non–cellular material resulting in cell lysis and death (Cobo et al., 2005; Dev et.al., 2011).
Further, if culture is contaminated by mycoplasma, the organisms have the capability to alter the morphology, function, metabolism, and growth and attachment cells to the culture vessel (McGarrity, 1976). Apart from these, presence of chemical contaminants (impure media, serum or organic compounds) in the cell culture media uncleansed storage vessel, pipettes, glassware due to malfunction in the sterilization procedures may cause unfavorable effects to the cells (Freshney, 2010). As far as selection and isolation of cells from tissue or organ is concerned, the contamination risk is low only removed aseptically. But in general, external tissues and organs pose a greater risk of contamination whereas internal organs are considered sterile with the exception of the digestive tract (Lester, 2007).
To prevent bacteria and/or fungi contaminations during cell culture, apart from following aseptic techniques and good laboratory practices, supplementation of the culture media with antibiotics and/or antimycotics is often done. A wide range of suitable preparations are available from relatively specific antibiotics, e.g., penicillin/ streptomycin solutions, to broader spectrum antibacterial/ antimycotic agents such as kanamycin or amphotericin B (Martinez–Liarte et al., 1995). However, the antibiotics chosen should not to be toxic to the cells in culture and their application and concentration may be decided depending on the type of contamination experienced in the individual laboratory (Lelong–Rebel et al., 2009). But higher doses of antibiotics could alter the phenotype or genotype of the cells and their prolonged use often cause negative impact on cultured cells affecting the cellular DNA as well as protein synthesis process which in turn interfere with cell metabolic processes (Kuhlmann, 1995; Toullec, 1999)
The culture media should have optimum range of pH, oxygen, osmolality, temperature and adequate buffering system to support good growth and proliferation of cultured animal cells.
Animal cells placed in a nutritionally complete tissue culture medium grow well when the medium is buffered at a pH in the range 7.2 to 7.4 (Williamson and Cox, 1967). However, a slight variation in pH level is essential depending on the type and origin of cells. For instance, some normal fibroblast cells require pH of 7.4 – 7.7, whereas transformed cells do better at pH 7.0 – 7.4 (Eagle, 1973). Although, Eisinger et al (1979) reported that at pH 5.5, epidermal cells could be maintained, but this level has not been universally adopted. On the other hand, most cultured cells stop growing when pH falls from 7.0 to 6.5 and start their losing viability between pH 6.0–6.5. In such cases, the medium should be changed (Freshney, 2010). To overcome this problem, it is important to measure the exact pH of the culture media. Further, as most culture media are buffered using bicarbonate, the pH level of the medium can be checked by a pH indicator like phenol red. Accordingly, the media may be changed / replenished, if its color turns yellow (acid) or purple (alkali) during cell culture. However, it is advisable not to use media with phenol red for studies using serum–free media formulations, as it is reported to interfere with the sodium potassium homeostasis. To neutralize its effect, serum or bovine pituitary hormone may be included in the culture medium (Karmiol, 2000).
Oxygen plays an important role in the physiology of cells by regulating their function, differentiation, and survival. Although cultured cells depend on the dissolved oxygen obtained from the surrounding medium to maintain metabolic activities, it is sparingly soluble in cell culture media. At higher concentration, dissolved oxygen inhibits the growth and metabolism of a number of cell types and may be toxic to cells due to the generation of free radicals. Additions of selenium and free radicals scavengers (glutathione) are reported to reduce oxygen toxicity (McKeehan et al., 1976). Further, proliferation of cells lowers the dissolved oxygen concentration in culture media, which in turn drops the pH due to release of lactic acid as a by–product of cell metabolism (Hanson et al., 2007). From this it is clear that too much medium inhibits oxygen diffusion resulting in slow cell growth where as too little may lead to cell death. As both dissolved oxygen and pH are good indicators of cell growth, their optimum concentration or level should be maintained throughout the culture period as the rate of cellular growth and metabolism are strongly dependent on these two factors (Miller et al., 1988).
Osmolarity is an important process variable during in vitro cultivation of mammalian cells Cell structure and function can be influenced by the osmolality of the cell culture medium. Therefore, maintaining an appropriate osmolality in the culture medium is required for proper growth and function of cells (Kruse and Paterson, 1973). It should be similar to the osmolality of the natural environment of the animal cells, in the range of 260–320mosmol/kg. Once an osmolality is selected for a medium, it must be maintained at that level (+/–10mosmol/kg), as the change in osmolality of culture medium causes detrimental effects on cell, either delay the cell growth or accelerate its cell death (Freshney, 2010). In case of any change in osmolality of culture medium due to addition of acids, bases to the medium; it should be measured using osmometer and adjusted accordingly.
Like pH, optimum temperature (37 0C) is required for the proper growth of the animal cells. However, the incubation temperature depends on the species and type of cells being cultivated, as cells from certain cultures like skin may require lower temperatures (Unchern, 1999). At lower temperatures, although the growth is arrested, cells can survive at as low as 4 °C temperature. Lowering the culture temperature of cells reduces glucose and glutamine consumption, proteolytic degradation rates and improves their tolerance against shear stress (Chen et al., 2004). At higher incubation temperature (>37 0C), cultured cells lose their viability (Khaparde and Roychoudhury, 2012). The enzymes of the cell at such temperature denature or become inactive whereas at low temperature the CO2 concentration increases decreasing pH level (Freshney, 2010), thereby influencing growth of cells. A consistency in the temperature of cultured animal cells is to be maintained to get desired results.
The purpose of a buffer in cell culture is to maintain pH within a very narrow range and resist changes in pH in when small amounts of acid or base are added. The buffers to be used in cell cultures must possess distinctive characteristics like high water solubility and minimal organic solvents solubility. Further, they should not permeate into cell membranes, exhibit any toxic effects towards cells and have pKa values at or near physiological pH (Mohan, 2003).The most commonly used buffer in tissue culture media is the bicarbonate and phosphate systems but in some cases, tris buffers are also in use (Eagle, 1971).
Amongst these, bicarbonate buffer offers advantages because of its low toxicity, low cost and nutritional benefits to the culture and maintains pH near 7.4 (Freshney, 2010). Apart from maintaining pH, bicarbonate buffers also helps in glucose metabolism and proper functioning of ion and acid–base transporters (Bonarius et al., 1995; Kanaan et al., 2007). But buffers when used with in vitro biological systems have their advantages and disadvantages. For example, cell culture using bicarbonate buffer (pKa of 6.3 at 37 0C) needs to be maintained at an appropriate temperature and gas mixture (typically, 37 0C, 5–10% CO2) in a cell incubator. On the other hand, phosphates in phosphate buffer (pKa, 6.9 at 37 0C) form insoluble complexes and inhibit the activity of some enzymes whereas tris buffer (pKa of 7.9 at 37 0C is reported to be cytotoxic at higher concentrations (Williamson and Pox, 1967). As each medium has a recommended CO2 tension and buffer concentration to maintain correct pH and osmolality, buffers should be tested for side effects, if any, on cells types before use (Coutu and Schroeder, 2013). To overcome the problems, a new range of zwitterionic buffers like N–tris (hydroxymethyl) methyl–2–aminoethanesulphonic acid (TES), N–2–hydroxyethylpiperazine–N'–ethanesulphonic acid (HEPES) has been developed which exhibit low interference with biological processes. These buffers, covering a pKa range from 6 to 8 are very soluble, have low binding capacities for divalent cations and are stable (Good et al., I966). Amongst these, HEPES has superior buffering capacity (pKa = 7.3 at 37 0C) and does not require a controlled gaseous atmosphere (Shipman, 1969). But it is toxic to some cell types at higher concentrations and relatively expensive (Zigler et al., 1985). Further, media containing HEPES can be photoactivated, if exposed to normal fluorescent lighting, producing hydrogen peroxide and free radicals that are toxic to cells (Wang, 1976; MacMichael, 1986).
Attachment of cells to culture vessels is a critical step for in vitro culture of primary cells (Freshney, 2010). This is because most normal cells and explants are anchorage–dependent and therefore, attachment to a substrate is required for their replication and subsequent growth (Folkman and Moscona, 1978; Danen and Yamada, 2001). Although glass, because of its optical properties and surface charge, was the substrate or matrix of choice during early days, it has now been replaced by synthetic plastic, usually polystyrene. But due to its hydrophobic nature, these materials do not provide a suitable surface for cell attachment (Curtis et al., 1983). To overcome the problem, now days, these materials are treated either through wet chemical processing, plasma treatment (glow discharge, corona, and flame) or radiation treatment (ultraviolet radiation and laser treatment) to produce a charged wettable surface (Guruvenket et al., 2003).
Considerable progress has also been made for identification and characterization of specific matrix proteins (glycoproteins) like collagen, gelatin, fibronectin, laminin and serum spreading factors (vitronectin) that promote and/or influence in vitro cell attachment to the surface or substratum of the culture vessel to induce adhesion and rapid spreading of cells (Damsky et al., 1984; Crouch et al., 1987). These materials improve the adhesive properties of the culture flask bottom to which negatively charged cells attach (Freshney, 2010). This helps to produce an extra cellular matrix, facilitates attachment, proliferation and further spreading of the cells (Kleinman et al., 1987). Apart from these, the substrate can also be conditioned by treating with serum, plasma or spent medium obtained from the same or another culture (Stampfer et al., 1980). Conditioning the surface of the culture dish with biocompatible materials like Silicon nitride also facilitate growth of cells (Medina Benavente et al., 2014). However, the culture substrate or other biomaterials used as substrate should be compatible and not exert any adversely effect on the cells through leaching of toxin (Laluppa et al., 1997).
In recent times, there is a growing interest for culture of cells in three dimensional (3–D) system within a polymeric matrix (also termed scaffold) as these materials mimic the in vivo conditions and increase cell–cell interactions compared to normal two dimensional cell culture (Abbott, 2003; Harrington et al., 2013). Various synthetic biodegradable polymers including poly–lactic acid (PLA), polyglycolide (PG), and their co–polymers are most widely used in the fabrication of polymeric scaffolds (Mikos et al., 1993; Mikos et al., 1994a, Mikos et al., 1994b; Carletti et al., 2011). Amongst these, polymeric scaffold materials (PLA) blended with natural polymers like alginate, gelatin and dextran have been explored with success for adhesion, attachment and proliferation of fibroblast cells in primary culture by Nanda et al (2014).
Growth factors are special polypeptides with stimulating effects on growth and division of certain types of cells. They act as signals and facilitate cell proliferation and differentiation mechanism (Tingjun et al., 2003). Cells from multicellular organisms are dependent upon exogenous signals. But growth factors are present in negligible amount in the nutrient medium (nanogram and pictogram level per millilitre of serum). So, they have to be added in the media for survival, growth, and proliferation to promote in vitro proliferation and differentiation of specialized cells. Over the years, considerable attention has been devoted to the role of peptide growth factors in the regulation of poultry and mammalian cell growth (Cyrino and Mulvaney, 1999). Notable major peptide growth factors (GF) are fibroblast GF, epidermal GF, platelet–derived GF, vascular Endothelial GF, Insulin GF (IGF–1, IGF–2) and transforming GF (Florini and Magri, 1989; Ewton and Florini, 1990; Florini et al., 1991). These growth factors are mostly used in serum–free media and act either individually or synergistically with each other, or with other factors like proteins (bovine serum albumin), polyamines, hormones (insulin and hydrocortisone) and nutrients (cholin, linolic acid etc.) to promote in vitro proliferation and differentiation of cells (Freshney, 2010).
Although media, nutrients, physico–chemical, growth & attachment factors play an important role in establishment of cell culture and subsequent subcultures; these may or may not give rise to an established cell line. It is because of the fact that normal cells in primary culture do not proliferate indefinitely but instead are mortal and can only divide a limited number of times and die out after a fixed number of population doubling. This is known as the Hayflick limit. Often referred to as crisis, the cells enter the state of senescence whereby cells move from actively dividing to a non–dividing state (Bols and Lee, 1994; Crane, 1999). This stage is determined by a number of intrinsic factors regulating cell cycle, such as pRb and p53 (Munger and Howley, 2002), and is accompanied by shortening of the telomeres as almost all cells and tissues, with the exception of post–mitotic cells show progressive shortening of telomeres with increased age (Wright and Shay, 2002; Ben–Porath and Weinberg, 2004). As a result, the protective ends of the chromosomes, the telomeres, gradually shorten with each cell cycle and when a critical telomere length is reached, the cell is unable to divide and the cell enters an irreversible state of quiescence (Barker et al., 2000). This is thought to involve the tumor suppressor gene p53, which arrests cell cycle progression (Kuilman et al., 2010). Cells that overcome the senescent state and have the potential to divide indefinitely in culture are referred to as immortalised (Crane, 1999). Therefore, for a cell line to become established or continuous, it must undergo transformation or immortalization. This can be induced through chemical mutagens, infection with a transforming virus or through transfection (Takarada et al., 1989; Bodnar et al., 1998; Guo et al., 2003; Butler and Nowak, 2004). Immortalization of cells or development of continuous cell line from normal cells requires telomere maintenance apart from optimal culture conditions as pointed out by several researchers (Mathon et al., 2001; Herbert et al., 2002; Wright and Shay, 2002). From this, it is clear that media, attachment factors, growth factors and other supplements simply optimize the conditions for cell proliferation and provide more opportunities to develop a continuous cell culture, but do not ensure development of a cell line from normal tissue.
Cell–culture Database : http://cell–lines.toku–e.com
Databases On Medicine And Molecular Biology: http://www.meddb.info
Cell Line Data Base (CLDB): (HyperCLDB) : http://bioinformatics.istge.it/cldb/cldb.php
Bio–Information Web A complete information of Web–resources of Biology : http://bioinfoweb.com/CLDB–list–Y.htm
American Type Culture Collection (ATCC): http://www.atcc.org
Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ): http://www.dsmz.de
European Collection of Cell Cultures (ECACC): http:// www. hpacultures.org.uk /collections /ecacc.jsp
Japanese Collection of Research Bioresources (JCRB): http://cellbank.nibio.go.jp
RIKEN Bioresource Center Cell Bank: http://www.brc.riken.go.jp/lab/cell/english/g
AltTox. Org Non–animal Methods for Toxicity Testing: http://www.alttox.org/ ttrc/ resources / data bases. html
REFERENCES
Abbott A (2003). Cell culture: Biology's new dimension. Nature 424: 870–872.
http://dx.doi.org/10.1038/424870a
PMid:12931155
Allen RE, Dodson MV, Luiten LS and Boxhorn LK (1985). A serum–free medium that supports the growth of cultured muscle satellite cells. In Vitro Cell Dev. Biol. 21(11): 636–640.
http://dx.doi.org/10.1007/BF02623296
PMid:3905759
Amemiya CT, Bickham JW and Gold JR (1984). A cell culture technique for chromosome preparation in cyprinid fishes. Copeia 1: 232–235.
http://dx.doi.org/10.2307/1445065
Arora M (2013). Cell culture media: A review. Mater. Meth. DOI http:// dx.doi.org /10.13070 /mm.en.3.175.
http://dx.doi.org/10.1177/0163443713495508
Barker KS, Qiniou SMA, Wilson MR, Bengten E, Stuge TBG, Warr W, Clem LW and Miller NW (2000). Telomerase expression and telomere length in immortal leukocyte lines from channel catfish. Dev. Comp. Immunol. 24: 583–595.
http://dx.doi.org/10.1016/S0145-305X(00)00021-5
Barnes D and Sato G (1980). Methods for growth of cultured cells in serum–free medium. Anal. Biochem. 102: 255–270.
http://dx.doi.org/10.1016/0003-2697(80)90151-7
Ben–Porath I and Weinberg RA (2004). When cells get stressed: an integrative view of cellular senescence. J. Clin. Invest. 113: 8–13.
http://dx.doi.org/10.1172/JCI200420663
http://dx.doi.org/10.1172/JCI20663
PMid:14702100 PMCid:PMC300889
Bettger WJ and McKeehan WL (1986). Mechanisms of cellular nutrition. Physiol. Rev. 66: 1–35.
PMid:3511479
Bhunia AK and Wampler JL (2005). Animal and cell culture models for foodborne bacterial pathogens. In: Foodborne Pathogens: Microbiology and Molecular Biology (Eds. Fratamico PM, Bhunia AK and Smith JL), Caister Academic, Norfolk, 15–32.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichsteiner S and Wright WE (1998). Extension of life–span by introduction of telomerase into normal human cells. Science 279: 349–352.
http://dx.doi.org/10.1126/science.279.5349.349
PMid:9454332
Bols NC (1991). Biotechnology and aquaculture: The role of cell cultures. Biotechnol. Adv. 9: 31–49.
http://dx.doi.org/10.1016/0734-9750(91)90403-I
Bols NC and Lee LEJ (1994). Cell lines: availability, propagation and isolation. In: Biochemistry and Molecular Biology of Fishes, Vol. 3, (Eds. Hochachka PW and Mommsen TP), Elsevier Science, Amsterdam, The Netherlands, 145–149.
Brunner D, Frank J, Appl H, Schoffl H, Pfaller W and Gstraunthaler, G (2010). Serum–free cell culture: The serum–free media interactive online database. Altex 27: 53–62.
PMid:20390239
Burgess D, Frerichs N and George S (1993). Control of metallothionein expression by hormones and stressors in cultured fish cell lines. Mar. Environ. Res. 35: 25–28.
http://dx.doi.org/10.1016/0141-1136(93)90008-N
Butler M (1991). The characteristics and growth of cultured cells. In: Mammalian Cell Biotechnology: A Practical Approach. Oxford University Press, New York, 1–26.
Butler M (2005). Animal cell cultures: recent achievements and perspectives in the production of biopharmaceuticals. Appl. Microbiol. Biotechnol. 68: 283–291.
http://dx.doi.org/10.1007/s00253-005-1980-8
PMid:15834715
Butler M and Jenkins H (1989). Nutritional aspects of the growth of animal cells in culture. J. Biotechnol. 12: 97–110.
http://dx.doi.org/10.1016/0168-1656(89)90009-6
Butler R and Nowak BF (2004). A dual enzyme method for the establishment of long and medium–term primary cultures of epithelial and fibroblastic cells from Atlantic Salmon gills. J. Fish Biol. 65: 1108–1125.
http://dx.doi.org/10.1111/j.0022-1112.2004.00521.x
Carere A, Stammati, A and Zucco F (2002). In vitro toxicology methods: impact on regulation from technical and scientific advancements. Toxicol. Lett. 127: 153–160.
http://dx.doi.org/10.1016/S0378-4274(01)00495-7
Carletti E, Motta A and Migliaresi C (2011). Scaffolds for tissue engineering and 3D cell culture. Meth. Mol. Biol. 695: 17–39.
http://dx.doi.org/10.1007/978-1-60761-984-0_2
PMid:21042963
Carrel A (1912). On the permanent life of tissues outside the organism. J. Exp. Med. 15: 516–528.
http://dx.doi.org/10.1084/jem.15.5.516
http://dx.doi.org/10.1084/jem.15.3.287
PMid:19867545 PMCid:PMC2124948
Carrel A and Burrows MT (1910). Cultivation of adult tissues and organs outside of the body. JAMA 55 (16): 1378–1381.
http://dx.doi.org/10.1001/jama.1910.04330160047018
Carrel A and Burrows MT (1911). Cultivation of tissues in vitro and it's technique. J. Exp. Med. 13: 386–396.
http://dx.doi.org/10.1084/jem.13.3.387
Castillo AA, Morier DL, Perez V and Mendez DC (1991). Use of goat serum as a substitute for calf serum for growing various primary cultures from vertebrates. Rev. Cubana Med. Trop. 43: 89–92.
Chaudhuri TK and Chakravarty AK (1983). Goat serum as a substitute for fetal calf serum in in vitro culture of murine lymphocytes. Indian J. Exp. Biol. 21: 494–496.
PMid:6674144
Chen Z, Wu B, Liu H, Liu X and Huang P (2004). Temperature shift as a process optimization step for the production of pro–urokinase by a recombinant chinese hamster ovary cell line in high–density perfusion culture. J. Biosci. Bioeng. 4: 239–243.
http://dx.doi.org/10.1016/S1389-1723(04)70198-X
Cheng Li–Lin, Bowser PR and Spitsbergen JM (1993). Development of cell cultures derived from lake trout liver and kidney in a hormone–supplemented, serum–reduced medium. J. Aquat. Anim. Health 5: 119–126.
http://dx.doi.org/10.1577/1548-8667(1993)005<0119:DOCCDF>2.3.CO;2
Cobo F, Stacey GN, Hunt C, Cabrera C, Nieto A, Montes R, Cortes JL, Catalina P, Barnie A and Concha A (2005). Microbiological control in stem cell banks: Approaches to standardization. Appl. Microbiol. Biotechnol. 68: 456–466.
http://dx.doi.org/10.1007/s00253-005-0062-2
PMid:16012832
Cooke MJ, Philips SR, Shah DSH Athey D, Lakey JH and Przyborksi SA (2008). Enhanced cell attachment using a novel cell culture surface presenting functional domains from extracellular matrix proteins. Cytotechnology 56 (2): 71–79.
http://dx.doi.org/10.1007/s10616-007-9119-7
PMid:19002844 PMCid:PMC2259265
Coutu DL and Schroeder T (2013). Probing cellular processes by long–term live imaging – historic problems and current solutions. J. Cell Sci. 126: 3805–3815.
http://dx.doi.org/10.1242/jcs.118349
PMid:23943879
Crane MStJ (1999). Mutagenesis and cell transformation in cell culture. Meth. Cell Sci. 21: 245–253.
http://dx.doi.org/10.1023/A:1009864111163
http://dx.doi.org/10.1023/A:1009861505170
PMid:10627679
Crouch EC, Stone KR, Bloch M and McDivitt RW (1987). Heterogeneity in the production of collagens and fibronectin by morphologically distinct clones of a human tumor cell line: Evidence for intratumoral diversity in matrix protein biosynthesis. Cancer Res. 47 (22): 6086–6092.
PMid:2822240
Curtis ASG, Forrester JV, Mcinnes, C and Lawrie F (1983). Adhesion of cells to polystyrene surfaces. J. Cell Biol. 97: 1500–1506.
http://dx.doi.org/10.1083/jcb.97.5.1500
PMid:6355120
Cyrino JE and Mulvaney DR (1999). Mitogenic activity of FBS, FFE, insulin like growth factor–1 and FGF on brown bullhead catfish cells–BB cell line. Rev. Bras. Biol. 59(3): 517–525.
http://dx.doi.org/10.1590/S0034-71081999000300017
PMid:10765463
Damsky CH, Knudsen KA and Buck CA (1984). Integral membrane proteins in cell–cell and cell–substratum adhesion. In: The Biology of Glycoproteins (Ed. Ivatt RJ), New York, Plenum Press, 1–64.
http://dx.doi.org/10.1007/978-1-4684-7464-0_1
Danen EH and Yamada KM (2001). Fibronectin, integrins, and growth control. J. Cell Physiol. 189: 1–13.
http://dx.doi.org/10.1002/jcp.1137
PMid:11573199
Davis JM (2002). Basic Cell Culture. A Practical Approach. 2nd Ed., Oxford University Press.
de Magalhães JP (2004). From cells to ageing: a review of models and mechanisms of cellular senescence and their impact on human ageing. Exp. Cell Res. 300(1): 1–10.
http://dx.doi.org/10.1016/j.yexcr.2004.07.006
PMid:15383309
Dev K, Giri SK, Kumar A, Khuttan A, Yadav A, Mandhan RP, Mohania D, Phuilia SK, Singh L, Kumar V, Kumar M and Gautam SK (2011). Studies of various microbial contaminants during culturing of amniotic fluid derived cells in buffalo (Bubalus bubalis). Int. J. Anim. Biotechnol. 1(1): 110–116.
Dils R (1984). Explants and disaggregated tissue preparations as model systems in nutritional research: Advantages and pitfalls. Proc. Nutri. Soc. 43: 133–140.
http://dx.doi.org/10.1079/PNS19840037
PMid:6089217
Dodson MV and Mathison BA (1988). Comparison of ovine and rat muscle–derived satellite cells: response to insulin. Tissue Cell 20: 909–918.
http://dx.doi.org/10.1016/0040-8166(88)90032-8
Duell BL, Cripps AW, Schembri MA and Ulett GC (2011). Epithelial cell co-culture models for studying infectious diseases: benefits and limitations. J. Biomed. Biotechnol. 852419, 1–9.
http://dx.doi.org/10.1155/2011/852419
PMid:22007147 PMCid:PMC3189631
Dulbecco R and Freeman G (1959). Plaque Production by the polyoma virus. Virology 8: 396–397.
http://dx.doi.org/10.1016/0042-6822(59)90043-1
Eagle H (1955). Nutrition needs of mammalian cells in tissue culture. Science 122: 501–504.
http://dx.doi.org/10.1126/science.122.3168.501
PMid:13255879
Eagle H (1959). Amino acid metabolism in mammalian cell cultures. Science 130: 432–437.
http://dx.doi.org/10.1126/science.130.3373.432
PMid:13675766
Eagle H (1971). Buffer combinations for mammalian cell culture. Science 174: 500–503.
http://dx.doi.org/10.1126/science.174.4008.500
PMid:5110427
Eagle H (1973). The effect of environmental pH on the growth of normal and malignant cells. J. Cell Physiol. 82: 1–8.
http://dx.doi.org/10.1002/jcp.1040820102
PMid:4354070
Ehrensvard G, Fischer A and Stjernholm R (1949). Protein metabolism of tissue cells in vitro, the chemical nature of some obligate factors of tissue cell nutrition. Acta Physiol. Scand. 18: 218–230.
http://dx.doi.org/10.1111/j.1748-1716.1949.tb00614.x
PMid:18148815
Eibl D, Eibl, R and Portner R (2009). Mammalian cell culture technology: An emerging field. In: Cell and Tissue Reaction Engineering: Principles and Practice (Eds: Eibl R, Portner R, Czermark P, Catapano G and Eibl D), Springer–Verlag Berlin Heidelberg, 1–6.
http://dx.doi.org/10.1007/978-3-540-68182-3
http://dx.doi.org/10.1007/978-3-540-68182-3_1
Eisinger M, Lee JS, Hefton JM, Darzykiewicz A, Chiao J W and Deharven E (1979). Human epidermal cell cultures–growth and differentiation in the absence of dermal components or medium supplements. Proc. Natl. Acad. Sci. USA 76: 5340.
http://dx.doi.org/10.1073/pnas.76.10.5340
PMid:291951 PMCid:PMC413138
Ewton DZ and Florini JR (1990). Effects of insulin like growth factors and transforming growth on the growth and differentiation of muscle cells in culture. PSEPM, 194: 76–80.
Figlewicz DA, Dong L, Mlodzienski M and Turcotte JC (2000). Culture models of neurodegenerative disease. Ann. N. Y. Acad. Sci. 919:106–118.
http://dx.doi.org/10.1111/j.1749-6632.2000.tb06873.x
PMid:11083103
Fisher A (1925). Tissue culture: Studies in experimental morphology and general physiology of tissue cells in vitro. William Heinemann, London.
PMid:17246289
Florini JR and Magri KA (1989). Effects of growth factors on myogenic differentiation. Am. J. Cell Physiol. 25: 701–711.
Florini JR, Ewton DZ and Magri KA (1991). Hormones, growth factors and myogenic differentiation. Annu. Rev. Physiol. 53: 201–216.
http://dx.doi.org/10.1146/annurev.ph.53.030191.001221
PMid:2042960
Folkman JC and Moscona A (1978). Role of cell shape in growth control. Nature (London), 273: 345–349.
http://dx.doi.org/10.1038/273345a0
PMid:661946
Freshney RI (2010). Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications, 6th Edition, Wiley–Blackwell, ISBN: 978–0–470–52812–9.
Garcia AN, Vogel SM, Komarova YA and Malik AB (2011). Permeability of endothelial barrier: cell culture and in vivo models. Meth. Mol. Biol. 763: 333–354.
http://dx.doi.org/10.1007/978-1-61779-191-8_23
PMid:21874463
Gille JJ and Joenje H (1992). Cell culture models for oxidative stress: superoxide and hydrogen peroxide versus normobaric hyperoxia. Mutat. Res. 275 (3–6): 405–414.
http://dx.doi.org/10.1016/0921-8734(92)90043-O
Good NE, Winget GD, Winter W, Connolly TN, Izawa S and Singh RMM (1966). Hydrogen ion buffers for biological research. Biochemistry 5: 467.
http://dx.doi.org/10.1021/bi00866a011
PMid:5942950
Guo H, Zhang S and Li H (2003). Spontaneous neoplastic transformation of the gill cell line FG–3907 from the Olive Flounder, Paralichthys olivaceus. N. Am. J. Fish. Aqua. 65(1): 44–48.
http://dx.doi.org/10.1577/1548-8454(2003)065<0044:SNTOTG>2.0.CO;2
Guruvenket S, Komath M, Vijayalakshmi SP, Raichur AM and Rao MG (2003).Wettability enhancement of polystyrene with electron cyclotron resonance plasma with argon. J. Appl. Polym. Sci. 90: 1618–1623.
http://dx.doi.org/10.1002/app.12816
Ham RG (1965). Clonal growth of mammalian cells in a chemically defined synthetic medium. Proc. Natl. Acad. Sci. 53:288.
http://dx.doi.org/10.1073/pnas.53.2.288
PMid:14294058 PMCid:PMC219509
Ham RG (1981). Cell growth requirements–the challenges we face. In: The Growth Requirements of Vertebrate Cells In Vitro, (Eds. Waymouth C, Ham RG and Chapple PJ), Cambridge University Press, Cambridge, 1–15.
Ham RG (1984). Formulation of basal nutrient media. In: Cell Culture Methods for Molecular and Cell Biology Vol. 1, (Eds. Barnes DW, Sirbasku DA and Sato GH), Allan R. Liss, New York, 2–21.
Hamilton W and Ham R (1977). Clonal growth of Chinese hamster cell lines in protein–free media. In Vitro 13: 537–547.
http://dx.doi.org/10.1007/BF02627849
PMid:562838
Hanson MA, Ge X, Kostov Y, Brorson KA, Moreira AR and Rao G (2007). Comparisons of optical pH and dissolved oxygen sensors with traditional electrochemical probes during mammalian cell culture. Biotechnol. Bioeng. (9): 833–841.
http://dx.doi.org/10.1002/bit.21320
PMid:17216654
Harrington H, Rose FRAJ, Aylott JW and Ghaemmaghami AM (2013). Self–reporting scaffolds for 3–Dimensional cell culture. J. Vis. Exp. 7(81): e50608.
Harrison R (1907). Observations on the living developing nerve fibre. Pro. Soc. Exp. Biol. Med. 4: 140–143.
http://dx.doi.org/10.3181/00379727-4-98
Hartung T (2007). Food for thought on cell culture. Altex 24: 143–147.
PMid:17926373
Hashizume T, Yoshitomi S, Asahi S,Matsumura S, Chatani F and Oda H (2009). In vitro micronucleus test in HepG2 transformants expressing a series of human cytochrome P450 isoforms with chemicals requiring metabolic activation. Mutat. Res. 677: 1–7.
http://dx.doi.org/10.1016/j.mrgentox.2009.03.009
PMid:19501186
Herbert B–S, Wright WE and Shay JW (2002). p16 (INK4a) inactivation is not required to immortalize human mammary epithelial cells. Oncogene 21: 7897–7900.
http://dx.doi.org/10.1038/sj.onc.1205902
PMid:12420227
Hetrick FM and Hedrick RP (1993). New viruses described in finfish from 1988–1922. In: Annual Review of Fish Diseases, (Eds. Faisal M and Hetrick FM), Pergamon Press, New York, 187–207.
Hightower LE and Renfro JL (1988). Recent applications of fish cell culture to biomedical research. J. Exp. Zool. 248: 290–302.
http://dx.doi.org/10.1002/jez.1402480307
PMid:3062124
Holmes RJ (1967). Preparation from human serum of an alpha–one protein which induces the immediate growth of unadapted cells. In Vitro J. Cell Biol. 32: 297–308.
http://dx.doi.org/10.1083/jcb.32.2.297
PMid:10976223 PMCid:PMC2107251
Inamdar NK and Borenstein JT (2011). Microfluidic cell culture models for tissue engineering. Curr. Opin. Biotechnol. 22(5): 681–689.
http://dx.doi.org/10.1016/j.copbio.2011.05.512
PMid:21723720
Karmiol S (2000). Development of serum free media. In: Animal Cell culture (Ed. Master JRW), 3rd ed. Oxford University Press.
Keay L (1975). Symposium on animal cell culture and its applications. Biotechnol. Bioeng. VOL. XVII: 625–627.
Khaparde SS and Roychoudhury PK (2012). Effect of low culture temperature on urokinase production in hollow fiber reactor. Appl. Biochem. Biotechnol. 168: 1655–1663.
http://dx.doi.org/10.1007/s12010-012-9886-2
PMid:22976853
Kleinman HK, Lukenbill–Edds L, Cannon FW and Sephel GC (1987). Use of extracellular matrix components for cell culture. Anal. Biochem. 166 (1): 1–13.
http://dx.doi.org/10.1016/0003-2697(87)90538-0
Kruse PF and Patterson MK (1973). Determination and survey of osmolality in culture media. In: Tissue Culture: Methods and Application. Academic Press, New York, 703–709.
Kuhlmann I (1995).The prophylactic use of antibiotics in cell culture. Cytotechnology 19(2): 95–105.
http://dx.doi.org/10.1007/BF00749764
PMid:22359010
Kuilman T, Michaloglou C, Mooi WJ and Peeper DS (2010). The essence of senescence. Genes Dev. 24: 2463–2479.
http://dx.doi.org/10.1101/gad.1971610
PMid:21078816 PMCid:PMC2975923
LaIuppa JA, McAdams TA, Papoutsakis. TE and Miller WM (1997). Culture materials affect ex vivo expansion of hematopoietic progenitor cells. Biomed. Mater. Res. 36: 347–359.
http://dx.doi.org/10.1002/(SICI)1097-4636(19970905)36:3<347::AID-JBM10>3.0.CO;2-B
Lane C, Pax R and Bennett J (1987). L–glutamine: an amino acid required for maintenance of the tegumental membrane potential of Schistosoma mansoni. Parasitology 94: 233–242.
http://dx.doi.org/10.1017/S0031182000053919
PMid:3588012
Lawson VA, Vella LJ, Stewart JD, Sharples RA, Klemm H, Machalek DM, Masters CL, Cappai R, Collins SJ and Hill AF (2008). Mouse–adapted sporadic human Creutzfeldt–Jakob disease prions propagate in cell culture. Inter. J. Biochem. Cell Biol. 40(12):2793–2801.
http://dx.doi.org/10.1016/j.biocel.2008.05.024
PMid:18590830
Leibovitz A (1963). The growth and maintenance of tissue–cell cultures in free gas exchange with the atmosphere. Am. J. Hyg. 78: 173–180.
PMid:14063721
Lelong–Rebel IH, Piemont Y, Fabre M and Rebel G (2009). Mycobacterium avium –intracellulare contamination of mammalian cell cultures. In Vitro Cell. Dev. Biol.–Anim. 45: 75–90.
http://dx.doi.org/10.1007/s11626-008-9143-8
PMid:18855078
Lester K (2007). Primary cell cultures from cod (Gadus morhua) and Atlantic halibut (Hippoglossus hippoglossus). M. Phil Thesis, Stirling University.
MacMichael GJ (1986). The adverse effects of UV and short wavelength visible radiation on tissue culture. American Biotechnology Laboratory.
Marrec–Croq Le F, Glaise D, Guguen–Guillouzo C, Chesne C, Guillouzo A, Boulo V and Dorange G (1999). Primary cultures of heart cells from the scallop Pecten Maximus (Mollusca–Bivalvia). In Vitro Cell. Dev. Biol.–Anim. 35: 289–295.
http://dx.doi.org/10.1007/s11626-999-0073-x
Martinez–Liarte JH, Solano F and Lozano JA (1995). Effect of penicillin–streptomycin and other antibiotics on melanogenic parameters in cultured B16/F10 melanoma cells. Pigment Cell Res. 8(2): 83–88.
http://dx.doi.org/10.1111/j.1600-0749.1995.tb00646.x
PMid:7659681
Mathon NF, Malcolm DS, Harrisingh MC, Cheng L and Lloyd AC (2001). Lack of replicative senescence in normal rodent glia. Science 291: 872–875.
http://dx.doi.org/10.1126/science.1056782
PMid:11157166
Mazia D, Schatten G and Sale W (1975). Adhesion of cells to surfaces coated with polylysine: Applications to electron microscopy. J. Cell Biol. 66(1): 198–200.
http://dx.doi.org/10.1083/jcb.66.1.198
PMid:1095595
Mcfarland DC (1992). Cell culture as a tool for the study of poultry skeletal muscle development. J. Nutr. 122: 818–829.
PMid:1371806
McGarrity GJ (1976). Spread and control of mycoplasmal infection of cell cultures. In Vitro Cell. Dev. Biol. 12: 643–647.
http://dx.doi.org/10.1007/BF02797464
McKeehan WL (1977). The effect of temperature during trypsin treatment on viability and multiplication potential of single normal human and chicken fibroblasts. Cell Biol. Int. Rep. 1: 335–343.
http://dx.doi.org/10.1016/0309-1651(77)90063-7
McKeehan WL, Hamilton WG and Ham RG (1976). Selenium is an essential trace nutrient for growth of WI–38 diploid human fibroblasts. Proc. Natl. Acad. USA 73:2023–2027.
http://dx.doi.org/10.1073/pnas.73.6.2023
Medina Benavente JJ, Mogami H, Sakurai T and Sawada K (2014). Evaluation of silicon nitride as a substrate for culture of PC12 cells: An interfacial model for functional studies in neurons. PLoS ONE 9(2): e90189.
http://dx.doi.org/10.1371/journal.pone.0090189
PMid:24587271 PMCid:PMC3937378
Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska–Lightowlers ZM, Dong L, Figlewicz DA and Shaw PJ (2002). Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125: 1522–1533.
http://dx.doi.org/10.1093/brain/awf167
PMid:12077002
Meyvantsson I and Beebe DJ (2008). Cell culture models in microfluidic systems. Annu. Rev. Anal. Chem. 1: 423–449.
http://dx.doi.org/10.1146/annurev.anchem.1.031207.113042
PMid:20636085
Mikos AG, Bao Y, Cima LG, Ingber DE, Vacanti JP and Langer R (1993). Preparation of poly (glycolic acid) bonded fiber structures for cell attachment and transplantation. J. Biomed. Mater. Res. 27: 183–189.
http://dx.doi.org/10.1002/jbm.820270207
PMid:8382203
Mikos AG, Thorsen AJ, Czerwonka LA, Bao Y, Langer R, Winslow DN and Vacanti JP (1994a). Preparation and characterization of poly (L–lactic acid) foams. Polymer 35: 1068–1077.
http://dx.doi.org/10.1016/0032-3861(94)90953-9
Mikos AG, Lyman MD, Freed LE and Langer R (1994b). Wetting of poly (lactic acid) and poly (dl–lactic–co–glycolic acid) foams for tissue culture. Biomaterials 15: 55–58.
http://dx.doi.org/10.1016/0142-9612(94)90197-X
Miller WM, Blanch HW and Wilke CR (1988). A kinetic analysis of hybridoma growth and metabolism in batch and continuous suspension culture: effect of nutrient concentration, dilution rate, and pH. Biotechnol. Bioeng. 32: 947–965.
http://dx.doi.org/10.1002/bit.260320803
PMid:18587813
Mohan C (2003). Buffers. A guide for the preparation and use of buffers in biological systems. EMD Biosciences Inc., San Diego, Caifornia, 13.
Morgan J, Morton H and Parker R (1950). Nutrition of animal cells in tissue culture; initial studies on a synthetic medium. Proc. Soc. Exp. Biol. Med. 73: 1–8.
http://dx.doi.org/10.3181/00379727-73-17557
PMid:15402504
Moscona A (1952). Cell suspensions from organ rudiments of the early chick embryo. Exp. Cell. Res. 3: 539.
http://dx.doi.org/10.1016/0014-4827(52)90077-3
Mótyán JA, Tóth F and Tőzsér J (2013). Research applications of proteolytic enzymes in molecular biology. Biomolecules 3: 923–942.
http://dx.doi.org/10.3390/biom3040923
PMid:24970197 PMCid:PMC4030975
Munger K and Howley PM (2002). Human papillomavirus immortalization and transformation functions. Virus Res. 89(2): 213–228.
http://dx.doi.org/10.1016/S0168-1702(02)00190-9
Nanda PK, Swain P, Nayak SK, Dash S, Routray P, Swain SK and Patra BC (2009). Goat serum as an alternative to establish cell culture from Indian Major Carp, Cirrhinus mrigala. In Vitro Cell Dev. Biol.– Anim. 45: 148–151.
http://dx.doi.org/10.1007/s11626-008-9160-7
PMid:19118441
Nanda PK, Swain P, Nayak SK, Mishra SS, Jayasankar P and Sahoo SK (2014). Use of polymeric scaffold for in vitro growth of fibroblast–like cells of Indian Major Carp, Cirrhinus mrigala. Adv. Anim. Vet. Sci. 2 (3): 177–182.
http://dx.doi.org/10.14737/journal.aavs/2014/2.3.177.182
Nema R and Khare S (2012). An animal cell culture: Advance technology for modern research. Adv. Biosci. Biotechnol. 3: 219–226.
http://dx.doi.org/10.4236/abb.2012.33030
Newsholme P, Procopio J, Lima MMR, Pithon–Curi TC and Curi R (2003). Glutamine and glutamate—their central role in cell metabolism and function. Cell Biochem. Funct. 21: 1–9.
http://dx.doi.org/10.1002/cbf.1003
PMid:12579515
Nicholson BL (1989). Fish cell culture: An update. In: Advances in Cell Culture, 7th Edition, (Eds. Maramorosch K and Sato GH), New York, Academic Press, 1–18.
PMid:2642391
Ostrander GK, Blair JB, Stark B A, Marley GM, Bales WD, Veltri RW, Hinton DE, Okihiro M, Ortego LS and Hawkins WE (1995). Long term primary culture of epithelial cells from rainbow trout (Oncorhynchus mykiss) liver. In Vitro Cell. Develop. Biol.– Anim. 31(5): 367–378.
http://dx.doi.org/10.1007/BF02634286
PMid:7543343
Paranjape S (2004). Goat serum: an alternative to fetal bovine serum in biomedical research. Indian J. Exp. Biol. 42(1): 26–35. PMid:15274477
Parker RC (1961). Methods of tissue culture, 3rd Edition, London, Pitman Medical, 47.
Pasieka A and Morgan J (1959). Glutamine metabolism of normal and malignant cells cultivated in synthetic media. Nature 183: 1201–1202.
http://dx.doi.org/10.1038/1831201a0
PMid:13657053
Peakman M, McNab GL, Heaton ND, Tan KC and Vergani D (1994). Development of techniques for obtaining monodispersed human islet cells. Transplantation 57: 384–393.
http://dx.doi.org/10.1097/00007890-199402150-00012
PMid:7509087
Pleskach VA, Kozhukharova IA, Artsybasheva IA, Golio TA, Tarunina MV and Filatova NA (1994). The cultivation features, growth characteristics and oncogenicity of serum free populations of transformed fibroblasts. Tsitologiia 36: 806–815.
PMid:7701611
Rous P and Jones FS (1916). A method for obtaining suspension of living cells from the fixed tissue, and for the plating out of the individual cells. J. Exp. Med. XXIII: 549–555.
http://dx.doi.org/10.1084/jem.23.4.549
Roux W (1885). Beitrage Zur Entwicklungsmechanik des Embryos. Z. Biol. 21: 411–526.
Salyers AA and Whitt DD (2002). Bacterial Pathogenesis: A molecular approach, 2nd edition, ASM, Washington, DC.
Schneider M, Windbergs M, Daum N, Loretz B, Collnot EM, Hansen S, Schaefer UF and Lehr CM (2013). Crossing biological barriers for advanced drug delivery. European J. Pharm. Biopharm. 84(2): 239–241.
http://dx.doi.org/10.1016/j.ejpb.2013.03.009
PMid:23531604
Shenoy M (2007). Culture Media. In: Animal Biotechnology. Laxmi Publications (P) Limited, Daryaganj, New Delhi, p: 10–18.
Shipman C (1969). Evaluation of 4–(2–hydroxyethyl)–1–piperazineëthanesulfonic acid (HEPES) as a tissue culture buffer. Proc. Soc. Exp. Biol. Med. 130: 305–310.
http://dx.doi.org/10.3181/00379727-130-33543
PMid:5762514
Smets LA, Homburg CH and Van Rooy H (1979). Selective effects of trypsinisation on established and tumor derived mouse 3T3 cells. Cell Biol. Int. Rep. 3: 107–111.
http://dx.doi.org/10.1016/0309-1651(79)90114-0
Stampfer M, Halcones RG and Hackett AJ (1980). Growth of normal human mammary cells in culture. In Vitro 16: 415–425.
http://dx.doi.org/10.1007/BF02618365
PMid:6993343
Stoll T, Muhlethaler K, von Stockar U and Marison I (1996). Systematic improvement of a chemically–defined protein–free medium for hybridoma growth and monoclonal antibody production. J. Biotechnol. 45:111–123.
http://dx.doi.org/10.1016/0168-1656(95)00153-0
Takarada Y, Ojima Y and Takai A (1989). Establishment of a fish cell line transformed by MNNG, RFM. Proc. Japan Acad. 65(B): 108–111.
Tingjun F, Lingyun J and Xiaofeng W (2003). Effects of basic fibroblastic growth factor and insulin–like growth factor on cultured cartilage cells from skate, Raja porasa. Chin. J. Oceanol. Limnol. 21 (4): 305–311.
http://dx.doi.org/10.1007/BF02860424
Toullec JY and Jean–yves (1999). Crustacean primary cell culture: a technical approach. Meth. Cell Sci. 21: 193–198.
http://dx.doi.org/10.1023/A:1009833924792
PMid:10627671
Trinchese F, Liu S, Ninan I, Puzzo D, Jacob JP and Arancio O (2004). Cell cultures from animal models of Alzheimer's disease as a tool for faster screening and testing of drug efficacy. J. Mol. Neurosci. 24(1): 15–21.
http://dx.doi.org/10.1385/JMN:24:1:015
Unchern S (1999). Basic techniques in animal cell culture. In: Drug Delivery System Workshop. Bangkok, Thailand, 1–30.
Vierck J, Byrne K, Mir PS and Dodson MV (2000). Ten commandments for preventing contamination of primary cell cultures. Meth. Cell Sci. 22: 33–41.
http://dx.doi.org/10.1023/A:1009873912078
http://dx.doi.org/10.1023/A:1009826012986
http://dx.doi.org/10.1023/A:1017581302281
PMid:10650333
Vincent AM, Stevens MJ, Backus C, McLean LL and Feldman EL (2005). Cell culture modeling to test therapies against hyperglycemia–mediated oxidative stress and injury. Antioxid. Redox Signal. 7 (11–12): 1494–1506.
http://dx.doi.org/10.1089/ars.2005.7.1494
PMid:16356113
Wang RJ (1976). Effect of room fluorescent light on the deterioration of tissue culture medium. In Vitro 12: 19–22.
http://dx.doi.org/10.1007/BF02832788
PMid:1244326
Whitford WG (2005). Supplementation of animal cell culture media. Bioprocess. Int. 3:S28–S36.
Williamson JD and Cox P (1967). Use of a new buffer in the culture of animal cells. J. Gen. Virol. 2: 309–312.
http://dx.doi.org/10.1099/0022-1317-2-2-309
Wolf K and Ahne W (1982). Fish Cell culture. In: Advances in Cell Culture Vol 2. (Ed. Maramorosch K), New York Academic Press, 305–328.
Wolf K and Quimby MC (1969). Fish cell and tissue culture. In: Fish Physiology Vol. 3, (Eds. Hora WS and Randall DJ), Academic Press, New York, 253–303.
Wolff K (1988). Fish Viruses and Fish Viral Diseases. Cornell University Press, Ithaca, New York, 476.
Wright WE and Shay JW (2002). Historical claims and current interpretations of replicative ageing. Nat. Biotechnol. 20: 682–688.
http://dx.doi.org/10.1038/nbt0702-682
PMid:12089552
Wu MH, Huang SB and Lee GB (2010). Microfluidic cell culture systems for drug research. Lab on a Chip 10(8): 939–956.
http://dx.doi.org/10.1039/b921695b
PMid:20358102
Yamada KM and Olden K (1978). Fibronectins: adhesive glycoproteins of cell surface and blood. Nature (London) 275: 179–184.
http://dx.doi.org/10.1038/275179a0
PMid:357987
Yoshitomi S, Ikemoto K, Takahashi J. Miki H, Namba M and Asahi S (2001). Establishment of the transformants expressing human cytochrome P450 subtypes in HepG2, and their applications on drug metabolism and toxicology. Toxicol. In Vitro 15: 245–256.
Zigler J, Lepe–Zuniga J, Vistica B and Gery I (1985). Analysis of the cytotoxic effects of light–exposed HEPES–containing culture medium. In Vitro Cell Dev. Biol. 21: 282–287.
http://dx.doi.org/10.1007/BF02620943
PMid:4019356
Zimmerman AM, Vierck JL, O̓Reilly BA and Dodson MV (2000). Formulation of a defined medium to maintain cell health and viability in vitro. Methods Cell Sci. 22: 43–49.
http://dx.doi.org/10.1023/A:1009832828007
PMid:10650334



