Advances in Animal and Veterinary Sciences
Research Article

Contribution of Amyloid β-Peptides To Acrylamide-Induced Toxicity In Rat Brain Tissue
Eda Özturan Özer*, Suna Türkoğlu
Department of Biochemistry, Faculty of Medicine, Başkent University, 06530 Ankara, Turkey.
Abstract | Acrylamide (ACR) is a well-known neurotoxic agent. The exact mechanism of neurotoxicity remains to be clarified. The following study aimed to evaluate the possible contribution of amyloid-β peptides to acrylamide-induced toxicity in rat brain tissue. Rats were randomly assigned to three groups, six rats in each group: control, low-dose (5mg/kg, i.p.) and high-dose (50 mg/kg, i.p.) acrylamide treated groups. In brain tissues, acetylcholinesterase activity (AChE) ,malondialdehyde (MDA), reduced glutathione (GSH), soluable amyloid β1-42 (Aβ1-42) peptide concentrations and also level of amyloid fibril formation (fibrillation) were analyzed. ACR treatment significantly reduced brain AChE activities in a dose-dependent manner (p<0.001). However, free Aβ1-42 concentrations and aggregation levels of amyloid fragments were significantly increased in brain tissues in low-dose-ACR treated group (p<0.01 and p<0.05 respectively). On the other hand, high-dose ACR treatment caused a significant decrease in tissue MDA and GSH levels (p<0.01). Our results have indicated that Aβ peptide-derived oxidative stress seemed to play a crucial role in ACR-induced neurotoxicity.
Keywords | Acrylamide, Oxidative stress, Amyloid-beta-peptide, Fibrillation, Neurotoxicity, Rat brain
Received | June 25, 2019; Accepted | August 19, 2019; Published | January 01, 2020
*Correspondence | Eda Özturan Özer, Department of Biochemistry, Faculty of Medicine, Başkent University, 06530 Ankara, Turkey; Email: eozer@baskent.edu.tr
Citation | Özer EO, Türkoğlu S (2019). Contribution of amyloid β-peptides to acrylamide-induced toxicity in rat brain tissue. Adv. Anim. Vet. Sci. 8(1): 54-60.
DOI | http://dx.doi.org/10.17582/journal.aavs/2020/8.1.54.60
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2020 Özer and Türkoğlu. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Acrylamide (ACR) is the water-soluble monomer of polyacrylamide which is mainly used in laboratories and various industrial processes such as flocculating agents for waste water treatment, petroleum, paper and cosmetic industry (Zamani et al., 2017). The main exposure route to acrylamide toxicity for humans is industrial occupation and smoking but evidences have revealed that diet may be considered as the another important ACR source for human and animals. ACR does not occur naturally in diets but however, can be generated from food components during heat treatment as a result of Maillard reaction condensation of the carbonyl group of reducing sugars with the nucleophilic amino group of the amino acids (especially asparagine and lysine residues) (Lambert et al., 2018). Generation of ACR by this reaction mainly depends on the food cooking and processing conditions, especially temperature (above 120-150 C) (Sassano et al., 2017; Lambert et al., 2018). ACR is a well-known neurotoxic agent in both humans and experimental animals which can show dose- and time-dependent deleterious effects, mainly degeneration, on central and peripheral nervous systems (Kopanska et al., 2018; Lopachin, 2004). Rodent studies have also suggested that ACR reveals genotoxic, mutagenic and carcinogenic effects (Lopachin, 2004). However, it has been reported that neurotoxicity is the only effect that has been observed in humans (Li et al., 2006). Existence of oxidative stress, alterations of enzyme activities, changes of neurotransmitter metabolisms and also signaling pathways were some of the reported features of ACR-induced toxicity in brain tissue (Crofton et al., 1996, Tian et al., 2015; Kopanska et al., 2018) however, the exact mechanism(s) of neurotoxicity remains to be elucidated (Tian et al., 2015; Zhao et al., 2017).
Amyloid β-peptides (mostly Aβ1-40 and Aβ1-42) are monomers which are derived from the amyloid precursor protein by the catalytic activities of β- and ɣ- secretases (Fedele et al., 2015). In generally, these monomers may form oligomers which may assembly to protofibrils (Lopachin and Gavin., 2012; Fedele et al., 2015). Fibrils which are generated from these protofibrils may be constructed as assemblies. Amyloid fibrils are ordered aggregates, -sheet structure, of normally soluble peptides. More than 20 different peptides/proteins have been identified in assemblies of the fibrils in vivo, e.g. acetylcholinesterase. Also, presence of cross-linked oligomers, such as amyloid β1-42, in brain tissues has been reported (Alvareez et al., 1998; Fedele et al., 2015).
Investigations had revealed that soluble forms of amyloid -peptides might be more toxic than amyloid aggregates depending on their cellular concentrations, i.e. nanomolar concentration (Roher et al., 2009; Fedele et al., 2015). Also, experiments had shown that amyloid β peptide-associated oxidative and nitrosative stress might lead to neuronal dysfunction and neurotoxicity (Tramutola et al., 2016; Cherignon et al., 2018).
The probable contribution of amyloid β peptide on acrylamide-derived neurotoxicity has not been studied previously. The following study aimed (a) to confirm the existence of neurotoxicity triggered by ACR, (b) to evaluate the presumable oxidative stress induced by sublethal doses of acrylamide treatments, (c) to clarify the the possible involment of amyloid- β peptides in acrylamide-induced toxicity in rat brain tissue.
MATERIALS and METHODS
Chemicals
All chemicals were obtained from Sigma-Aldrich ( St Louis, MO, USA), and Merck (Darmstadt, Germany). Aβ1-42 ELISA kit was purchased from SunRed-Bio (Shangai, China).
Animals and Treatments
Male albino rats of Wistar strain (n=18, 18 week old) weighing 220-290 g were obtained and housed in Başkent University, Medical and Surgical Experimental Research Center (temperature 20 ± 2oC, humidity 50 ± 10% and 12h light: 12h dark cycle). Rats were supplied with standard laboratory diet and tab water ad libitum. All experimental procedures involving animals were approved by Başkent University Institutional Review Board and Ethics Committee, Ankara, Turkey with the project number DA06/01.
Rats were assigned to three groups, as saline treated control group (CONT; n=6), 5mg/kg acrylamide treated group (GRP I; n=6) and 50 mg/kg acrylamide treated group (GRP II; n=6). All treatments were done intraperitoneally (i.p.) for five days and after 48 h of last treatment, rats were anesthetized i.p. with ketamine 50 mg/kg and xylazine 10 mg/kg. They were sacrificed by intracardiac puncture. After isolation of tissues, wet weights of brain tissues were recorded and were kept at -86C for biochemical analysis. All biochemical studies were performed in duplicate by using left hemisphere of brain.
Determination of Tissue Acetylcholinesterase Activity
Brain tissues were homogenized in 0.1 M potassium phosphate buffer pH 7.4 using all glass homogenizer and then centrifuged at 10000xg for 10 min at 40 C. Supernatants were used as an enzyme source. Acetylcholinesterase (AChE) activity was determined by the method of Ellman. (Ellman et al., 1961) , which is based on monitoring the rate of color complex formation at 412 nm for 5 min against a sample blank by using a spectrophotometer (Shimadzu UV-1601, Japan). Assays were carried out at 250 C in 84 mM potassium phosphate buffer pH 7.4, 0.1 mM 5, 5’-dithiobis-2-nitrobenzoic acid and 0.84 ml of enzyme source. Reaction was initiated by the addition 0.4 mM acetylthiocholine (AcSCh). Activity was defined as µmole AcSCh utilized /min/gram tissue (U/g).
Determination of Tissue Malondialdehyde and Reduced Glutathione Concentrations
Brain homogenates were prepared in ice-cold 0.15 M KCl (10%, w/v) using all-glass homogenizer for the determination of tissue MDA and GSH concentrations.
MDA levels were determined in brain homogenates according to the method of Buege and Aust (Buege and Aust, 1978). Two volumes of the thiobarbituric acid reagent was added to one volume of sample and mixture was incubated in a boiling water bath for 15 min. After cooling, centrifugation was achieved at 1000x g for 10 min. The absorbance was measured at 535 nm against a reagent blank by using a spectrophotometer (Shimadzu UV-1601, Japan). Concentrations were quantified by using molar extinction coefficient of 1.56 x 105 M-1cm-1. The results were expressed as nmole MDA/g tissue.
GSH levels were assayed in tissue homogenates according to the method of Ellman (Ellman., 1959). After deproteinization of the samples, Ellman’s color reagent was added into the supernatants and then the absorbance of generated color complex was measured immediately at 412 nm against a reagent blank with a spectrophotometer (Shimadzu UV-1601, Japan). By using GSH standard curve, concentrations were calculated and results were expressed as µmole GSH/g tissue.
Determination of Tissue Aβ1-42 Levels
Tissue Aβ1-42 levels were determined by using ELISA kit (SunRed-Bio, Cat No: 201-11-0094, Shangai, China) which is based on a solid phase sandwich enzyme assay. Brain homogenates were prepared according to the instructions of the kit.
Standards and samples were pipetted onto monoclonal antibody coated wells of microtiter strips and the assay was carried out as indicated in the instructions of the manufacturer.
The optical densities were measured at 450 nm by microplate reader (Bio-Tek Instruments, INC.ELX 800, USA). Quantitation was carried out by a standard curve and expressed as ng/g tissue.
Thioflavin T Analysis
Thioflavin T (ThT) is a traditional dye revealing hydrophobic and ionic characters which provide binding of ThT to various peptides, polypeptides and proteins. The cross -sheet structure, found in most amyloid proteins is a specific binding target of ThT (Khurana et al., 2005; Biancala and Koide, 2010). ThT fluorescence assay was conducted as an indication of amyloid fibril formation, fibrillation (Nilsson, 2004; Griffin et al., 2010). In briefly, brain tissues were homogenized gently in 100 mM sodium phosphate buffer, pH 7.4, using glass-glass homogenizer (10%, w/v). After centrifugation, the ThT fluorescence of supernatant was determined using 8 μM ThT in 100 mM sodium phosphate buffer, pH 7.4, using spectrofluorimeter (Shimadzu RF-5301, Japan) using excitation and emission wavelengths of 442 and 482 nm respectively. Fluorescence intensities were recorded and expressed as arbitrary unit (A.U.).
Statistical Analysis
Analyses were evaluated by with SPSS, Version 17.0 Software. Univariate analysis of variance (ANOVA) coupled with Duncan’s post-hoc test was performed. Data were expressed as means ± Standard error of mean (SEM) and p-values less than 0.05 were considered as statistically significant.
RESULTS
Analysis of brain AChE activities was performed as an evaluation of neurotoxicity. As shown in Figure 1, the sublethal doses of ACR treatment caused a decrease in the activities of AChE significantly in a dose-dependent manner (p<0.001). Considering the AChE activities of control (840.5 ± 45.77 U/g tissue) and ACR treated groups (low- and high-doses), crucial decline was detected in high-dose of ACR treated one (560.0 ± 14.81 U/g tissue), comparing to low-dose ACR treated group (717.33 ± 10.66 U/g tissue).
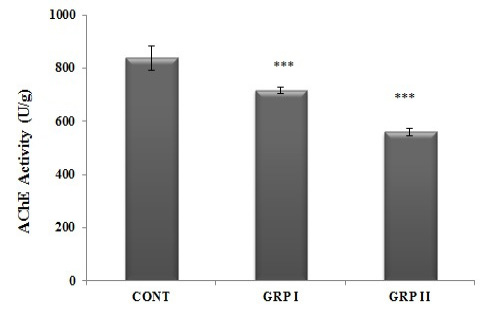
Figure 1: Brain AChE activities. Intraperitoneally five doses of saline (CONT), 5 mg/kg acrylamide (GRP I) and 50 mg/kg acrylamide (GRP II) injected groups. Treatments are detailed under experimental procedures section. Values are mean ± SEM. The sample size of each group is 6. *** p< 0.001; CONT vs GRP I vs GRP II
MDA concentrations were determined as a marker of lipid peroxidation index in brain tissues. ACR treatment altered MDA concentrations in a dose-related manner. As seen in Figure 2, in GRP II brain MDA concentrations significantly decreased when compared to both control and GRPI (31.0±1.64; 35.9±0.83; 37.4±0.62 nmole/g tissue, respectively; p<0.01).
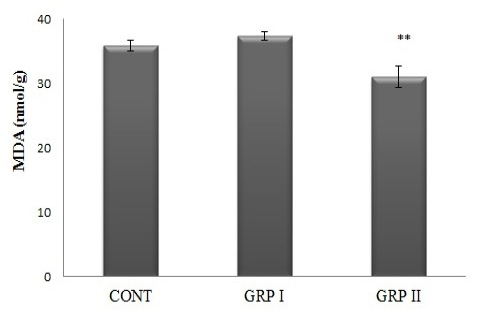
Figure 2: Brain MDA concentrations. Treatments are described in Fig 1. Values are mean ± SEM. The sample size of each group is 6. ** p<0.01; GRP II vs CONT and GRP I
Tissue GSH levels, as one of the parameters of redox status, were analized and data showed the similar alteration as seen in tissue MDA concentrations. Acrylamide treatment caused a significant decrease in brain GSH levels in the high dose- treated group (GRP II) comparing to control and low-dose ACR treated group, GRPI (5.5±0.30; 6.4±0.15; 6.7±0.11 mmole GSH/g tissue, respectively; p<0.01; Figure 3).
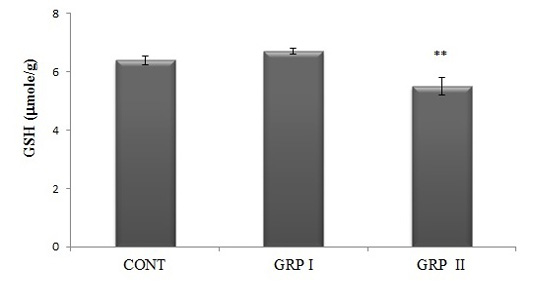
Figure 3: Brain GSH concentrations. Treatments are described in Fig.1. Values are mean ± SEM. The sample size of each group is 6. ** p<0.01; GRP II vs CONT and GRP I
As seen in the Table 1, ACR treatment altered tissue Aβ1-42 concentration in a dose-related manner. A significant increase in brain Aβ1-42 concentration was detected in GRP I when compared to control and high dose-ACR treated groups (p<0.01). The similiar stituation was observed again in terms of fibrillation in low-dose ACR treated group, thefibrillation was elevated significantly (p<0.05, Table 1).
Table 1: Effect of acrylamide treatment on brain Aβ1-42 concentrations and levels of fibrillation
| CONT | GRP I | GRP II | |
|
Aβ 1-42 (ng/g) |
11.4±0.13 |
18.5±0.17 ** |
12.6±0.12 |
| Fluorescence Intensity (A.U.) | 2.5±1.58 |
11.8±2.77 * |
2.0±1.26 |
Treatments are described in Figure 1. Values are mean ± SEM. The sample size of each group is 6.
* p<0.05; GRP I vs CONT and GRP II , ** p<0.01; GRP I vs CONT and GRP II
DIscussIon
In current experimental model, the validity of ACR-induced neurotoxicity was verified by two well defined evidences, i. e. decrease in body weight gain (Crofton et al., 1996; Lopachin et al., 2008; Ghareeb et al., 2010; Guano et al., 2018) and impairment of the cholinergic function, decrease in the activity of AChE (Antonio et al., 2003, Yousef and El-Demerdash., 2006).
Glutathione is one of the most important non-protein thiol in the brain tissue which has various functions in the central nervous system including neuroprotection (Antonio et al., 2003). In the body, detoxification of ACR will be achieved by enzymatic and nonenzymatic conjugation with GSH, which is the main route for detoxification in humans and experimental animals. Besides, metabolization of ACR via CYP2E1, minor route for detoxification, causes generation of a reactive epoxy compound, glycidamide, directly involved in mutagenic and carcinogenic effects of ACR, and also reactive oxygen species (ROS) (Wie et al., 2015, Sassano et al., 2017).
In the present study, ACR treatment caused the depletion of GSH concentration in the brain tissue. This situation occurred in a dose-related manner. Low-dose ACR treatment (5mg/kg) did not alter brain GSH concentrations, whereas high-dose acrylamide treatment (50mg/kg) caused a significant decrease in brain GSH concentration, below the basal level. Depletion of cellular GSH level in brain tissue may be due to enhancement of (a) conjugation reactions catalyzed by glutathione S-transferase, (b) formation of thiol-disulfide, GSSG, and/or protein (peptide)-glutathione disulfides (protein-SSG) (Tramutola et al., 2016). GSH depletion, whatever the reasons, can subsequently enhance the oxidative injury (Aoyama and Nakaki, 2012; Tramutola et al., 2016). It has been suggested that ACR intoxication produces ototoxicity, mediated by cellular glutathione depletion and adduction of protein cysteine residues (Lopachin et al., 2008).
Bioactive aldehydes, including MDA, are formed mostly during lipid peroxidation of polyunsaturated fatty acids of membrane phospholipids. Aldehydes of lipid peroxidation products are very reactive molecules having the ability to make aldehyde-aldehyde and aldehyde-protein/peptide products. Modification of several residues of peptides/proteins, especially lysyl residues, will be achieved by attacking MDA. They can be considered as toxic aldehydes due to the alterations of several cell functions. The presence of aldehyde-protein adducts had been demonstrated in a wide range of physiological and pathological conditions. Under physiological conditions, they can be found at low concentrations in tissues and plasma. They can participate in the control of certain biological processes, such as signal transduction, cell proliferation and differentiation (Haas and Salkoe, 2007) On the other hand, they can propagate and amplify oxidative injury. Besides intra- and intermolecular protein/peptide cross-links, MDA, itself, is prone to produce dimers, trimers and larger polymers by aldol condensation (Pizzimenti et al., 2013; Halliwell and Gutteridge., 2015).
In present experimental model, again low-dose ACR treatment (5mg/kg) did not alter brain MDA concentration, whereas high-dose acrylamide treatment (50mg/kg) caused a decrease in brain MDA concentration significantly. Modulation of MDA concentration, below the basal level, accompanying with GSH depletion indicated the existence of oxidative stress in the brain tissues of high-dose ACR treated group, whereas no apparent oxidative stress in terms of lipid peroxidation and intracellular antioxidant status, GSH level, in the brain tissues of low-dose ACR treated group. Reduction of brain MDA levels in high-dose ACR treated group may reveal the probable existence of aldehyde-aldehyde and aldehyde-protein/peptide products which causes the alteration of cellular functions.
In current model, studied the possible incorporation of amyloid β-peptides into ACR-induced toxicity in brain tissue, which would be the first dealing with this concept. Present data have indicated that amyloid β-peptides (especially A1-42 ) seemed to play crucial roles in ACR-induced toxicity in brain tissue.
In the present study, low-dose ACR treatment significantly enhanced the concentration of soluble A1-42 and also the level of fibrillation of amyloid β-peptides. Besides, brain tissue AChE activity decreased significantly. On the other hand, as mentioned before, there was no sign of oxidative stress in terms of lipid peroxidation and intracellular redox status, GSH level. Also, there was no significant alteration in body weight gain.
Current data have suggested that a significant elevation of A1-42 peptide may be a response of brain tissue against exogenous toxic aldehyde itself, i.e. ACR, indicating a sign of probable neurotoxicity, e.g. decrease in body weight gain, and oxidative stress. On the other hand, significant decrease in brain AChE activity might be accepted as an early sign of ACR-induced neurotoxicity (Ghareeb et al., 2010; Fedele et al., 2015).
Mechanism(s) of elevation of A1-42 peptide had not been explained but it had been attributed to elevation of A1-42 peptide may be accepted as an acute phase response of brain tissue against toxic aldehyde (Fedele et al., 2015).
Besides significant elevation of brain A1-42 peptide concentration, as mentioned above, low-dose ACR treatment also caused to an increment of the level of fibrillation of Aβ peptides level in the brain tissue.
Soluble Aβ oligomers are correlated significantly with neuronal dysfunction, damage and also pathological symptoms. Also cross-linked oligomers, e.g. amyloid β1-42, could be detected in biological samples (Bruggink et al., 2013; Sengupta et al., 2016). It had been postulated that excessive soluble amyloid fibers are inactivated into fibrillar core structures surrounded by glial cells and there might be a continuous dynamic structure between small, soluble oligomers and insoluble deposits. Also, soluble oligomers can bind to other macromolecules or to the cell membrane and can therefore become insoluble (Butterfield and Kanski, 1999; Roher et al., 2009; Bruggink et al., 2013). It had been revealed that brain AChE forms stable complexes with Aβ peptide during assembly and AChE-amyloid complexes lead to enhance the toxicity (Alvarez et al., 1998).
In present experimental model, elevation of fibrillation of Aβ peptides may be considered as a defense mechanism against toxic monomers or oligomers in low-dose ACR treatment, since no oxidative stress and neurotoxicity have been detected apparently. However, there was no significant modulation of soluble A1-42 concentration and the fibrillation of amyloid β-peptide level in high-dose acrylamide treated rat brain tissues, although there was an existence of oxidative stress and neurotoxicity.
Studies had indicated that methionine-derived sulfuramyl radicals in amyloid peptides are responsible for the initiation of oxidative stress such as Aβ-derived lipid peroxidation, mitochondrial dysfunction, neuroinflammation and apoptosis (Butterfield and Kanski., 1999). Lipid peroxidation products can react directly with Aβ peptides, producing peptide/protein adducts, which exacerbates the formation of oligomers and insoluble aggregates (Chen et al., 2007). AChE is one of the defined core protein which contributes aggregation procedure. In vitro experiments have also indicated that MDA significantly enhances not only the beta-sheet formation of amyloid peptides, but also the aggregation of them at various stages by modification of fibrils, such as formation of protofibrils and aggregates (Ellman., 1959; Chen et al., 2007). Therefore, enhancement of oxidative stress causes Aβ oligomerization and toxicity (Liu et al., 2008; Pizzimenti et al., 2013).
In the present study, incorporation of aldehyde modified- Aβ peptides and also aldehyde-aldehyde adducts into the fibrillation process may probably be resulted irregular form(s) of fibrillation or regular forms with subtle changes in the dye, ThT, binding regions (Krebs et al., 2005). These situations presumably can be resulted as basal levels of A1-42 and also fibrillation.
In conclusion, current experimental model has indicated that administration of sublethal low-dose ACR causes to the elevation of Aβ peptides which will presumably be an acute-phase response of the organism. ACR-induced exacerbation of Aβ peptide formation resulted as enhancement of fibrillation process, which was evaluated as a kind of defense mechanism against stress/trauma. As exposure to exogenous aldehyde increased, sublethal high-dose, ACR-induced Aβ peptide-derived oxidative stress probably contributed to the toxicity in various aspects. Reduced MDA concentrations with depletion of GSH levels revealed the existence of oxidative stress and neurotoxicity became apparent.
Involvement of GSH depletion in neurodegeneration has been studied previously and it had been reported that GSH depletion was the primary cause of neurodegeneration and not only a result of oxidative stress (Halliwell and Guttridge., 2015). Neurons are more vulnerable to oxidative stress than glial cells, due to having less sufficient defense system including low GSH content. Improvement of brain GSH level may clarify the molecular mechanism of ACR-induced neurotoxicity, considering Aβ peptide-derived oxidative stress which may provide new aspects for neurodegenerative diseases in therapeutic approach.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
FUNDING STATEMENT
This study was financially supported by Başkent University Research Fund with the project number DA06/01.
Acknowledgements
The authors acknowledge to Baskent University Research Fund for supporting this work.
authors contribution
Eda Özturan Özer: Designed the present study, performed all experiments and calculations, analyzed data and prepared tables and figures. Suna Türkoğlu: Designed the present study, evaluated the data and wrote the manuscript.
References



