Advances in Animal and Veterinary Sciences
Review Article
Mechanisms of Stroke Induced Neuronal Death: Multiple Therapeutic Opportunities
Suresh L Mehta*, Raghu Vemuganti
Department of Neurological Surgery, University of Wisconsin, Madison WI 53792 USA.
Abstract | Stroke is one of the major contributors of mortality and morbidity worldwide. It results due to sudden blockade of blood flow into the brain thereby leading to breakdown of hemodynamic, biochemical and metabolic processes in the affected territory. These alterations are dependent on the severity and duration of the ischemia and modulated by pathophysiologic mechanisms that include excitotoxicity, calcium overload, oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction, acidosis, inflammation and activation of cell death pathways. As there is no current therapy to minimize post-stroke brain damage and most of the clinical trials failed, it is imperative to design novel approaches including glutamate receptor antagonists, free radical scavengers/anti-oxidants, anti-inflammatory and anti-apoptotic agents, and growth factors to prevent the progression and increase the plasticity of neuronal damage after stroke. Additionally, rigorous in vitro and in vivo animal testing of potential drugs molecules is necessary to reduce the failure of clinical trials.
Editor | Kuldeep Dhama, Indian Veterinary Research Institute, Uttar Pradesh, India.
Received | September 11, 2014; Revised | September 13, 2014; Accepted | September 13, 2014; Published | September 16, 2014
*Correspondence | Suresh L Mehta, Department of Neurological Surgery, USA; Email: mehta@neurosurgery.wisc.edu
Citation |Mehta SL, Vemuganti R (2014). Mechanisms of stroke induced neuronal death: multiple therapeutic opportunities. Adv. Anim. Vet. Sci. 2 (8): 438-446.
DOI | http://dx.doi.org/10.14737/journal.aavs/2014/2.8.438.446
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2014 Mehta and Vemuganti. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Stroke is characterized as a sudden blockade of blood flow to the brain, thereby depriving the brain tissue of oxygen and glucose supply. This results in the breakdown of metabolic processes in the affected territory. Stroke is one of the leading causes of death and disability worldwide and according to the WHO, 15 million people suffer stroke worldwide each year. Out of which, 5 million die and another 5 million are permanently disabled. High blood pressure is one of the significant contributors (>12.7 million) of stroke. In the United States, more than 140,000 people die each year from stroke, whereas stroke death rate each year averages approximately 650,000 in Europe. Although, the incidence of stroke is declining in developed countries, largely due to efforts to lower blood pressure and reduce smoking, however, the overall rate of stroke remains high due to the aging of the population (The internet stroke centre). Stroke is classified as ischemic (~80%) and hemorrhagic including subarachnoid hemorrhage (20%). Ischemic stroke is induced as a result of cardiac arrest or focal loss of blood flow to the brain due to atherosclerotic or embolic blockade of an artery. Hemorrhagic stroke on contrary occurs when the loss of blood flow caused due to rupture of blood vessels on or within the brain. The consequence of a stroke is the massive tissue infarction and damage to neurons due to activation of complex pathophysiological mechanisms based upon the severity, duration, and location of the reduced blood supply. Damage is sometime delayed in some of the regions of the brain depending on the vulnerability of the neurons; however it eventually leads to the loss of brain function (Heiss et al., 1999; Hermann et al., 2001). Since, most of the strokes are of ischemic in nature and widely studied, this brief review will discuss some of the major changes that follows during Ischemic stroke.
Ischemia do not affects the brain tissue equally due to differential reduction in blood flow in affected territory. Therefore, severe ischemia occurs in the central zone (core), where blood supply will be reduced to <7mL/100g/min (Bandera et al., 2006) and neuronal damage in this region is irreversible due to uncontrolled necrotic cell death. The core region is surrounded by a zone of borderline ischemic tissue called ischemic penumbra with blood flow ranging from 7-17mL/100g/min (Baron, 1999). Due to the dynamic bioenergetic disturbances in this topographic locus, the neurons in the penumbra remains metabolically active and potentially salvageable, although electrically silent (Ginsberg and Pulsinelli, 1994; Baron, 1999; Hata et al., 2000). During ischemia, the energy level in the infarct core is severely impaired with near zero level in the centre and a gradient toward the periphery. Energy level in the penumbra is maintained because of retrograde perfusion from the adjacent arteries (Bang et al., 2011; Jung et al., 2013). Penumbra can be identified by multitracer positron emission tomography or perfusion-weighted imaging/diffusion-weighted imaging mismatch in acute stroke (Reid et al., 2012), and can also be described in molecular terms, such as changes in heat shock proteins 70, which is selectively induced in response to transient ischemic stroke in the area of decreased blood flow and oxygen delivery (Hata et al., 2000; Sharp et al., 2000). Hence, penumbral tissue has the potential for recovery and is the target for therapeutic intervention. However, the only therapy approved for the ischemic stroke is the recombinant tissue plasminogen activator, with certain limitations such as narrow therapeutic time window of three to six hours and risk of haemorrhage (Hickenbottom and Barsan, 2000). Disappointingly, most of the clinical trials ended prematurely due to disruption of normal brain functions and adverse effects, which points out the essential need to understand the complexities of hemodynamic, biochemical, and neurophysiological alterations responsible for reducing the viability of neurons following stroke (Mehta et al., 2007).
It is also important to mention here that in order to comprehend the complicated pathophysiological cascades of molecular and cellular events, various cell lines and animal models have been developed to mimic clinical stroke conditions. The importance of these models lies in revealing the potential damaging mechanisms and preclinical testing of drugs designed for neuroprotection that ultimately might improve functional recovery after stroke. This review will briefly discuss some of the pathophysiological mechanisms that can be targeted to improve neurological outcome (Figures 1 and 2) and various models developed to study cerebral stroke.
Targeted mechanisms to improve neurological outcome
Glutamate Excitotoxicity and
Calcium Overload
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS), acting through both ligand gated ion channels (ionotropic receptors), N-methyl-D-aspartate (NMDA) receptor, 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl) proprionate receptor, and kainate receptor subtypes, and G-protein coupled (metabotropic) receptors. Activation of these receptors plays an important role in the basal excitatory synaptic transmission and many forms of synaptic plasticity such as long-term potentiation and long-term depression through calcium influx (ionotropic) and internal calcium mobilization (metabotropic). Pharmacological studies in rodents and recent clinical studies in humans have shown that the extracellular concentration of glutamate is raised to levels toxic to neurons during ischemic stroke (Hagberg et al., 1985; Benveniste, 2009; Lai et al., 2014). In addition, the reduced uptake and metabolism of glutamate by glial cells may also be responsible in part for the increased extracellular glutamate during ischemia (Zanchin et al., 1995).
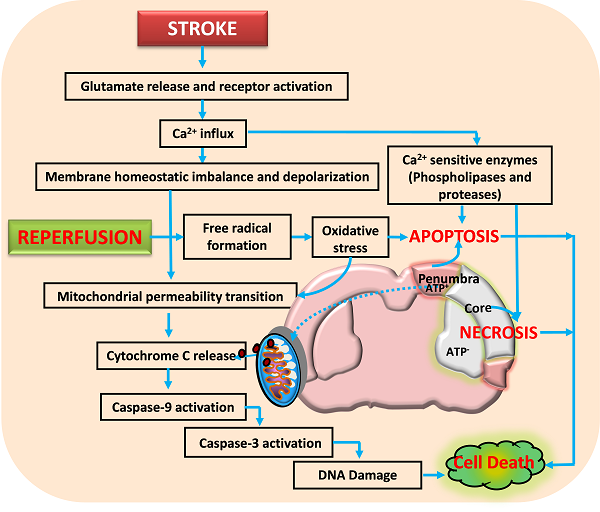
Activation of glutamate receptors following ischemic stroke leads to excitotoxicity and calcium influx. This impairs the neuronal homeostasis leading to activation of several calcium dependent pathways that include proteases and nucleases. Reperfusion aggravates the neuronal damage by forming free radicals that damage the membranes, proteins and DNA. Further, the opening of mitochondrial permeability transition pore releases various proapoptotic molecules including cytochrome C that activate apoptotic cell death. Neuronal death following ischemia will be a result of necrotic, apoptotic and necroptotic mechanisms depending on the severity of insult.
Neurons control both intracellular calcium levels and the location of calcium ions through influx, efflux, buffering, and storage. However, excessive calcium influx through glutamate receptors and release from intracellular stores during ischemic stroke exceeds the capacity of calcium regulatory mechanisms, thereby leading to activation of calcium-dependent proteases, lipases, phosphatases, and endonucleases that either directly damage cell structure or induce mitochondri-al permeability transition (MPT) pore, formation of oxygen reactive species (ROS) and cell death (Arundine and Tymianski, 2003; Bano and Nicotera, 2007; Lai et al., 2014). Thus, glutamate receptors are considered as potential therapeutic targets (figure 2) for CNS disorders such as stroke and traumatic brain injury, or chronic degenerative diseases including amylotrophic lateral sclerosis, multiple sclerosis, Huntington’s disease, Parkinson’s disease and Alzheimer’s disease.
Oxidative Stress
Brain is a highly metabolically active organ. Being only 2-3% of total body, it receives 15% of cardiac output, consumes about 20% of the body’s oxygen and 15% of total body glucose. ROS are the important by-product during cellular metabolic activities and are constantly detoxified/scavenged by various cellular antioxidant enzymes. However, it has been demonstrated that ischemia increases the formation of reactive oxygen species (ROS), thereby leading to oxidative stress and damage to proteins, lipids, and DNA in ischemic tissues (Chan, 2001; Rodrigo et al, 2013). This ultimately results in the death of neurons. Also, free fatty acid (FFA) metabolism produces oxygen free radicals and products of FFA metabolism like arachidonic acid, thromboxane and leukotrienes that can promote cerebral edema, produce vasoconstriction, induce platelet aggregation and play a role in ischemic brain damage. It has been increasingly reported that reducing the oxidative stress through various approaches including the application of antioxidant therapy have beneficial protective effect against ischemic stroke by mechanisms that involve inhibition of mitochondrial dysfunction and apoptosis (Imai et al., 2003; Warner et al., 2004; Rodrigo et al., 2013.
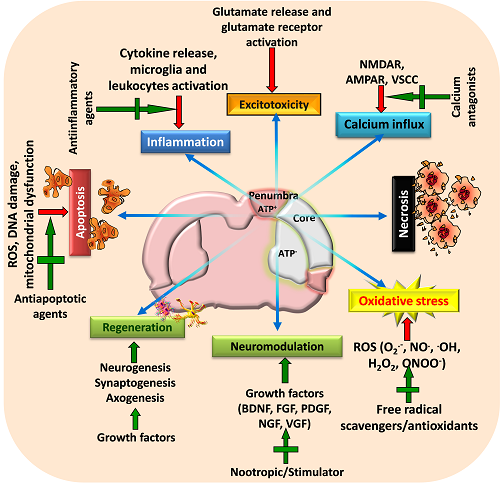
Figure 2: A schematic view of various targeted neuroprotective approaches for minimizing post-stroke brain damage
Stroke activates various pathophysiological mechanisms including glutamate excitotoxicity, oxidative stress, and inflammation that impair neuronal survival and results in neuronal death through necrosis, apoptosis or necroptosis. Moreover, stroke also impairs neuromodulation and regeneration of cells. Therefore, strategies that can target these pathways as well as restore growth factors and initiate regeneration of neurons may provide beneficial outcome following stroke. Adapted and modified from Mehta et al. (2009).
Inflammation
Inflammation plays an important role in the pathogenesis of ischemic stroke and acute brain injuries (Denes et al., 2010). The major players involved in the inflammatory cascades are cytokines (IL-1, IL-6, TNF-α, and TGF-β), adhesion molecules (selectins, integrins, and immunoglobulins), eicasanoids and inducible neuronal nitric oxide synthase, which are produced within hours after the onset of ischemia and continue for days (Jin et al., 2010). Initiation of inflammatory reactions contribute to irreversible brain damage and involve interplay between endothelial cells, astrocytes, microglial cells and leukocytes (granulocytes, monocyte/macrophages and lymphocytes).
In addition, oxidative stress triggers the expression of a number of pro-inflammatory genes through transcription factors like NF-κB including cytokines and adhesion molecules. Likewise, mitogen activated kinases, p38, ERK 1/2 and SAPK/JNK are also involved in the regulation of expression of inflammatory genes including IL-6, iNOS, TNF-ɑ and IL-1β, and extracellular-matrix-related genes (MMP9 and MMP13) following ischemic stroke (Pun et al., 2009; Vikman et al., 2007). Therefore, it is strongly believed that targeting inflammatory process by various approaches including inhibition of leukocyte infiltration, selective inflammatory molecule antagonists, antibody based targeted approach, inhibiting MAPKs dependent stress signalling and antioxidant treatment may reduce the brain damage and improve stroke outcome (Figure 2).
Cell Death Pathways
It has been increasingly established that mitochondria, besides providing majority of the energy for most of the cellular and molecular functions, plays a major role in ROS production, calcium homeostasis and regulation of cell death (necrosis/apoptosis) during cerebral ischemia. It acts as a reservoir for a multitude of apoptogenic proteins (Ferrer et al., 2003; Nakka et al., 2008; Niizuma et al., 2010). As mentioned above neurons in the ischemic core die through necrotic cell death whereas apoptotic cell death is the major contributor of the neuronal damage in ischemic penumbra. Necrosis occurs in an indiscriminate fashion through osmotic homeostatic imbalance, rupture of the plasma membrane and subsequent leakage of cellular contents into the surrounding tissue, which invariably results into inflammatory response. Apoptosis on the other hand is a controlled form of cell death and is classified as mitochondrial mediated (intrinsic) or death receptor mediated (extrinsic) forms. Its initiation depend upon the stimulus and mode of activation such as mitochondrial injury, intracellular calcium elevation, oxidative stress and tumour necrosis factor activation, growth factor deficiency, DNA damage and p53 induction. Numerous studies have shown that mitochondrial dysfunction is one of the major events during ischemic stroke and other neurodegenerative diseases (Murakami et al., 1998; Solenski et al., 2002; Li et al., 2012). For example, mitochondrial uptake of calcium has been considered as one of the important step in restricting calcium fluctuation. However, following stroke excessive intracellular calcium increase through glutamate receptor activation particularly NMDA receptor can cause mitochondrial ultra-structural alterations and calcium-dependent opening of mitochondrial permeability transition pore (Castilho et al, 1998; Kristian and Siesjo, 1998; Stout et al., 1998; Szydlowska and Tymianski, 2010). MPT allows the release of soluble proteins such as cytochrome c, apoptosis-inducing factor, and Smac/Diablo, etc. (Szydlowska and Tymianski, 2010; Stout et al., 1998).
Mitochondrial dysfunction and mitochondria-initiated apoptosis are involved in pathophysiology of cerebral ischemia (Christophe and Nicolas, 2006). The early release of cytochrome c into the cytosol regulates the activation of caspase-9 which further activates caspase-3 that activates calpains, caspase-activated endonuclease CAD/DFF40 (Stennicke and Salvesen, 1998; MacManus and Buchan, 2000), and promotes the cleavage of DNA repair enzyme PARP, with an ensuing breakdown of nuclear structure (Krupinski et al., 2000; Eliasson et al., 1997). Thus, mitochondria dysfunction and mitochondrial-initiated cell death pathway plays an important role in mediating ischemic brain damage. A third from of cell death called necroptosis is reported by Degterev et al. (2005) based on the view that neuronal cell death caused by ischemic injury contains a substantial non-apoptotic and non-necrosis component. It is initiated by death receptors under conditions that are not favourable to apoptosis and is represented by biochemical and ultra-structural features of apoptotic and necrotic pathways in the same cell (van Lookeren Campagne and Gill, 1996; Unal-Cevik et al., 2004; Wei et al., 2004), which appears to be a backup mechanism to eliminate the damaged cells in the brain. Therefore, therapeutic intervention during initial hours after reversible neuronal injury (Figure 2) through various approaches including caspases inhibitors, anti-apoptotic Bcl-2 proteins, and necroptosis inhibitor (necrostatin-1) etc. have shown promising result in reducing neurological deficit and brain damage after stroke. Thus, targeting cell death mechanisms offer an opportunity to reduce/prevent the progression of neuronal damage.
Modelling of stroke
Various in vitro and in vivo models have been developed to mimic clinical stroke. The overall goal of using these models is to effectively understand the pathophysiological mechanisms, testing novel compounds with therapeutic potential and to translate the outcomes more effectively to clinical settings. These model systems, therefore, can be used to screen molecules with thrombolytic, neuroprotective and neurorestorative potential.
In vitro Models
In vitro ischemic models are important part of stroke studies. The effect of ischemia in vitro can be visualized using cell lines, primary cell cultures and brain slice cultures. Neuronal cells including HT22 (murine hippocampal neuronal cell line), PC12 (a pheochromocytoma of the rat adrenal medulla derived cell line), SH-SY5Y (human neuroblastoma cell line) are some of the cell lines used to study ischemia pathology. Additionally, primary cells derived from cerebral capillary endothelium or from various brain tissue such as cortex, hippocampus, striatum and cerebellum are used to study the effects of ischemia. Moreover, stroke related changes can also be studied in vitro using organotypic cortical or hippocampal brain slices cultures (Frantseva et al., 1999; Cho et al., 2007; Wang and Andreasson, 2010). The cultured brain slices have advantages over cell line cultures as they retains many essential organizational features of the host tissue including neuronal connectivity. Likewise, genetic and pharmacological manipulations are easier to perform in cell free systems (Cho et al., 2007). The various model approaches used to mimic stroke in vitro are oxygen-glucose deprivation, hypoxia or chemical hypoxia through 3-nitropropionic acid (3-NPA), cyanide (CN) and deoxyglucose, glutamate induced excitotoxicity etc.
In vivo Models
Besides in vitro models, in vivo models have been developed in various animal species including mice, rats, gerbils, rabbits, cats, dogs, monkeys and baboons (Casals et al., 2011). Rat and mouse strains are widely used for stroke related studies due to their close resemblance to human cerebrovascular anatomy and physiology, easy handling, homogeneity in various strains and importantly reproducibility of stroke damage. In addition, genetic manipulations especially in mouse has special advantage to understand the complexity of the mechanisms involved in damage progression. However, higher animals are also used due to their closer resemblance to humans in terms of behaviour and sensorimotor function.
Animal models developed are classified as focal or global, complete or incomplete and transient or permanent ischemia. The focal ischemic model involves the occlusion of the middle cerebral artery (MCA) either transiently or permanently, mimicking ischemic stroke in humans (Longa et al., 1989; Garcia 1984; Belayev et al., 1996; Traystman, 2003). The global model is more relevant to human cardiac arrest and involves bilateral occlusion of the carotid and vertebral arteries for a short duration. For inducing focal ischemia, MCA can be occluded with an intraluminal suture (Longa et al., 1989; Belayev et al., 1996) or with a vascular clip either transiently or permanently. Focal ischemic stroke can also be produced by one of the following; injecting a homologous small clot or fibrin-rich autologous clots (Orset et al., 2007), collagen (Purdy et al., 1989), viscous silicone (Lauer et al., 2002), polyvinylsiloxane (Yang et al., 2002), retractable silver ball (Molnár et al., 1988) and heterologous atheroemboli (Rapp et al., 2003), photoactive dye Rose Bengal (Watson et al., 1985) and endothelin-1 (Macrae et al., 1993). The importance of the occlusion model also lies in the fact that reperfusion injury in humans may be due to lysis of a thrombo-embolic clot. In addition to focal ischemic stroke model, the global model can be produced by two-vessel occlusion with transient bilateral common carotid artery (CCA) occlusions plus hypotension (Smith et al., 1984), four-vessel occlusion (4-VO) by transient bilateral CCA occlusions plus permanent vertebral artery occlusions (Pulsinelli and Brierley, 1979), decapitation without recirculation (Ginsberg and Busto, 1989), neck cuff inflation (Nemoto et al., 1977), ventricular fibrillation (Todd et al., 1981), cardiopulmonary resuscitation (Berkowitz et al., 1991) to study the various aspects of brain damage. The global ischemia leads to the selective neuronal necrosis, whereas focal cerebral ischemia is more likely to develop infarction with pan necrosis. Gerbils are generally preferred to study the ischemic effects on hippocampal area as they lack the posterior communicating arteries necessary to complete the circle of Willis in rats and humans.
Conclusions
Stroke is one of the major contributors of mortality and morbidity worldwide. Many studies have convincingly shown that brain damage during stroke is mediated by accumulation of extracellular glutamate, calcium overload, free radical generation and mitochondrial dysfunction, inflammation and cell death activation. Approaches that target these mechanisms seem to have potential to develop therapy, reduce the brain damage and improve neurological outcome following stroke and related brain injuries. The understanding of the pathophysiological complexity of these mechanisms is achieved by the use of various in vitro and in vivo models that are developed to mimic human stroke conditions, and test potential antistroke agents.
References



