Advances in Animal and Veterinary Sciences
Research Article

Cloning and Optimizing the Culture Parameters for Expression of R1 and R2 Repeat Regions of P97 Adhesin from Mycoplasma hyopneumoniae in Escherichia coli
Phung Thang Long*, Le Quoc Viet, Le Viet Quan, Dong Huu Rin, Nguyen Xuan Hoa, Le Duc Thao, Le Dinh Phung, Nguyen Thi Thu Hien, Dinh Thi Bich Lan
Faculty of Animal Sciences and Veterinary Medicine, University of Agriculture and Forestry, Hue University, Hue 530000, Vietnam
Abstract | Mycoplasma hyopneumoniae (M. hyopneumoniae) is the causative agent of porcine enzootic pneumonia (PEP), a chronic respiratory disease that affects pigs of all ages worldwide and causes considerable economic losses in the pig industry. The R1 and R2 repeat regions of P97 adhesin (P97R1R2) of M. hyopneumoniae play important roles in adherences to the host cells to initiate the infection process, and are capable to confer immunogenicity, and potential candidates for development of the recombinant subunit vaccines. The objective of this study was to clone P97R1R2 gene fragment of M. hyopneumoniae isolated from pigs in Thua Thien Hue province, Vietnam and optimize culture parameters (cultivation temperature, culture media, inducer concentration, induction time and incubation time) for improving expression of recombinant P97R1R2 protein in E. coli. The result showed that the nucleotide sequence of P97R1R2 gene fragment of M. hyopneumoniae was 779 bp in length, corresponding to 272 amino acids with 10 repeats of AAKPE(V) in R1 and 3 repeats of GS(A) PN (S) QGKKAE in R2. Expression of the P97R1R2 in E. coli BL21 StarTM (DE3) produced a fusion protein with molecular weights of approximately 33.42 kDa (including 3.7 kDa of fusion fragment of pET-200/D-TOPO vector). Furthermore, the cultivation temperature at 25oC, YJ medium, Isopropulβ-D-1-thiogalactopyranoside (IPTG) concentration of 0.6 mM, induction time at an optical density of 1.5 at 600 nm, and post-induction incubation time for 6 h were determined to be the optimal for the expression of target protein. In conclusion, this study was successful in cloning P97R1R2 gene fragment of M. hyopneumoniae isolated from pigs in Thua Thien Hue province, Vietnam and optimizing selected culture parameters for higher expression of recombinant P97R1R2 protein in E. coli BL21 StarTM (DE3) cells.
Keywords | Mycoplasma hyopneumoniae, Porcine enzootic pneumonia, P97R1R2 gene fragment, Expression of P97R1R2 protein, Recombinant protein
Received | August 14, 2019; Accepted | October 10, 2019; Published | November 22, 2019
*Correspondence | Phung Thang Long, Faculty of Animal Sciences and Veterinary Medicine, University of Agriculture and Forestry, Hue University, Hue 530000, Vietnam; Email: thanglong@huaf.edu.vn
Citation | Long PT, Viet LQ, Quan LV, Rin DH, Hoa NX, Thao LD, Phung LD, Hien NTT and Lan DTB (2019). Cloning and optimizing the culture parameters for expression of R1 and R2 repeat regions of P97 adhesin from Mycoplasma hyopneumoniae in Escherichia coli. Adv. Anim. Vet. Sci. 7(12): 1067-1075.
DOI | http://dx.doi.org/10.17582/journal.aavs/2019/7.12.1067.1075
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2019 Long et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Mycoplasma hyopneumoniae (M. hyopneumoniae) is the causative agent of porcine enzootic pneumonia (PEP), a chronic respiratory disease that affects pigs of all ages worldwide (Austen et al., 2006), resulting in retarded growth and reduced feed efficiency, and causes considerable economic losses in the pig industry (Sheldrake et al., 1991; Thaker et al., 1998). Pigs with M. hyopneumoniae infection are predisposed to potentially fatal secondary infections by the porcine reproductive and respiratory syndrome virus (Thaker et al., 1999), and influenza viruses (Jang and Kim, 2007) as well as other opportunistic bacterial pathogens (Thaker et al., 1998; Calsamiglia et al., 1999). Vaccination is the most cost-effective strategy for controlling and prevention of PEP (Maes et al., 2007; Holst et al., 2015). The currently used commercial PEP vaccines mainly are inactivated and attenuated live vaccines, and their efficacy has not yet fully elucidated (Tao et al., 2019).
P97 of M. hyopneumoniae was the first adhesin identified. It is highly conserved among different M. hyopneumoniae strains (Zhang et al., 1995; Wilton et al., 1998), and is best-defined potential protective antigen against M. hyopneumoniae (Zhang et al., 1995; Simionatto et al., 2013). P97 contains two repeat regions, R1 and R2, located on the C-terminus portion (Hsu et al., 1997). The R1 with 9-15 the AAKPV(E) repeats (Wilton et al., 1998) participates in the adherence of M. hyopneumoniae to the cilia of pigs respiratory epithelium (Hsu et al., 1997; Minion et al., 2000; Deutscher et al., 2010). The R2 with 3-5 repeats of GS(A)PN(S)QGKKAE involves in the attachment of M. hyopneumoniae to the extracellular matrix of the respiratory tract (Jenkins et al., 2006). Both R1 and R2 repeat regions are needed to bind heparin in the pathogesesis of M. hyopneumoniae (Jenkins et al., 2006; Deutscher et al., 2010). Recently, much effort has been made to develop more effective vaccines against M. hyopneumoniae based on both R1 and R2 repeat regions and through recombinant DNA techniques (Shimoji et al., 2003; Ogawa et al., 2009; Okamba et al., 2010; Lee et al., 2014). In fact, in order to produce recombinant antigen proteins, the bacterium E. coli is the most popular host used due to its well-known genetics, high growth rates on inexpensive media, and a wide range of possible expression vectors (Terpe, 2006). However, the production efficacy of recombinant proteins in E. coli is dependent on culture conditions (Malik et al., 2016; Mühlmann et al., 2017).
To our knowledge, there is very few study of cloning and optimizing culture parameters for expression of both R1 and R2 repeat regions of M. hyopneumoniae P97 adhesin in E. coli, including Vietnam. Therefore, the objective of this study was to clone P97R1R2 gene fragment of M. hyopneumoniae isolated from pigs in Thua Thien Hue province, Vietnam and optimize culture parameters consisted of cultivation temperature, culture media, IPTG inducer concentration, induction time and incubation time for improving expression of recombinant P97R1R2 protein in E. coli BL21 StarTM (DE3) to contribute to the development of new subunit vaccines against M. hyopneumoniae.
MATERIALS AND METHODS
Tissue sample collection, plasmids and culture media
The fresh lung tissue samples of pigs suffering from PEP originating from unvaccinated herds were collected, immediately frozen in liquid nitrogen, and stored at - 80oC until use. The pGEM®-T Easy vector (Promega, USA) was used as cloning system. The pET200/D-TOPO® vector (Invitrogen, USA) harboring the T7 promoter allows expression of the recombinant protein. E. coli TOP10 (Invitrogen, USA) and E. coli BL21(DE3) strains (Invitrogen, USA) were used as host strains for plasmid DNA amplification and production of recombinant protein, respectively. The five different culture media of LB (Loc et al., 2013), YJ (Loc et al., 2011), TB (Shen et al., 2007), HSG (Miksch et al., 2008) and modified M9ZB (Lee et al., 2000) were used for expression of target recombinant protein.
DNA isolation and PCR amplification of P97R1R2 gene fragment
Genomic DNA of M. hyopneumoniae was isolated by Phenol-Choloroform extraction method and used as a template in PCR amplification with P97R1R2 forward primer: 5’-CACCATGGCAAAATTAGACGATAATCT-3’ and P97R1R2 reverse primer: 5’-GCGCATTTTTACCTTGTTTTTTAA-3’, which designed based on the coding DNA sequence of the P97R1R2 gene fragment (accession no. AY512905 on the GenBank). To enable directional cloning in the further, the forward primer contains the four nucleotides of CACC at the 5’ end to base pair with the GTGG overhang sequence in pET200/D-TOPO vector. For PCR amplification, a 25-µl PCR reaction mixture consisted of 1 µl DNA template (50 ng/µl), 12.5 µl 2×PCR master mix (2.4 mM dNTP, 0.3 units Taq DNA polymerase) (Fermentas, Canada), 1 µl each primer (10 pmol/µl) and distilled water was prepared, and a thermocycler (iCycler thermocycler, Bio-Rad, Hercules, CA) was used. The reaction mixture was then subjected to pre-denaturation step at 95oC for 5 min, followed by 30 cycles of which each included a denaturation step at 95ºC for 45 sec, an annealing step at 47oC for 1 min, and an extension step at 72oC for 1 min. After a final elongation step at 72oC for 10 min, PCR product was tested by 1% (w/v) agarose gel electrophoresis and stained with 0.5 µg/L ethidium bromide solution for 15 min. Electrophoresis image was analyzed by DyNA Light, Dual Intensity UV Transilluminator (Labnet).
Cloning and sequencing of the P97R1R2 gene fragment
The PCR product was purified from the agarose gel using Isolate II PCR and Gel Kit (Bioline, USA), then was inserted into pGEM®-T Easy vector according to TA cloning method. A 10-µl ligation mixture consisted of 50 ng the vector (50 ng/µl), 5 µl buffer, 3 units T4 DNA ligase (3 units/µl), 39 ng PCR product (39 ng/µl) and distilled water was prepared and incubated at 25oC for 1 h, and at 4oC overnight, then transformed into competent E. coli TOP10 cells by heat-shock method. The recombinant transformed cells were streaked on LB plate with 100 µg/ml ampicillin, 100 mM IPTG and 20 mg/ml X-gal for selection of recombinant clones. The colonies were screened and confirmed by colony direct PCR with M13 primer pairs of the pGEM®-T Easy vector. A positive colony was cultured on 5 ml LB medium supplemented with 50 µg/ml ampicillin for biomass production. Recombinant pGEM®-T Easy/P97R1R2 vector was then extracted by EZ-10 Spin Column Plasmid DNA MiniPreps Kit, BS614 (Bio Basic INC). PCR product was sequenced by the method of fluorescent dideoxy terminator on the 3130 Genetic Analyzer (Applied Biosystems®).
Expression and purification of recombinant P97R1R2 protein
The PCR product from recombinant pGEM®-T Easy vector was cut out from 1% agarose gel and purified using ISOLATE II PCR and Gel Kit, and then was ligated into pET200/D-TOPO® expression vector harboring promoter T7. A 6-µl ligation mixture consisted of 1 µl of 20 ng vector, 1 µl of salt solution, 2 µl of 8 ng PCR product and distilled water was prepared and incubated at 25oC for 1 h. Recombinant pET200/D-TOPO® vector was then transformed into E. coli StarTM BL21(DE3) cells according to the manufacture’s instruction. The recombinant transformed cells were streaked on LB plate with 100 µg/ml kanamycin and incubated at 37oC overnight for the selection of recombinant clones. A positive colony was cultured on 20 ml LB medium containing 100 µg/ml kanamycin and incubated at 37oC in a rotary shaker at 200 rpm to an optical density at 600 nm (OD600) = 0.8. IPTG was added to a final concentration of 0.8 mM for induction, and the IPTG-induced culture was further incubated at 37oC, 150 rpm for 8 h post-induction. The cell biomass was centrifuged at 10000 rpm 4oC for 10 min, and the pellet was resuspended in appropriate volume of TNE buffer (50 mM Tris-HCl, 100 mM NaCl, 2 mM EDTA, pH 7.4) containing 1% Triton X-100, 0.008 mg/ml lysozyme and kept on ice-water bath for 1 h. After gentle sonication for 5 min, the culture was centrifuged at 10000 rpm 4oC for 5 min, and the supernatant and pellets were collected for identification of target protein localization. Inclusion body proteins in the pellets were solubilized with 8 M urea buffer (8 M urea, 0.01 M Tris, 0.1 M NaH2PO4, pH 8.0) in a rotary shaker at 150 rpm, 30oC for 2 h, then centrifuged at 10000 rpm for 15 min for collecting the supernatant. In the parallel experiment, the transformed E. coli BL21 (DE3) cells were cultivated in the identical conditions, but without IPTG induction. An equal volume from each sample was mixed with sample loading buffer, heated at 100 oC for 10 min and analyzed 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
The recombinant P97R1R2 proteins were purified by ProBondTM Purification System Kit (Invitrogen, USA) according to the manufacturer’s instructions and loaded on 15% SDS-PAGE. The gel was then stained with Coomassie Blue R-250, and the image was analyzed by Quality One software (Ver 4.1, Bio-Rad).
Western blot analysis
The purified recombinant P97R1R2 proteins in SDS-PAGE gel were electrotransferred onto a nitrocellulose membrane (Bio-Rad) for 5 h and blocked with 3% skimmilk into phosphate-buffered saline (PBS) for 2 h. After washing three times for 5 min/each with PBS, the membrane was incubated with anti-His G-horseradish proxidase (HRP) antibody (Bio Rad) diluted with 3% skimmilk into PBS (diluted 1:2500) for 1 h. After washing six times for 5 min/each with PBS, target protein band was detected with 0.005% (w/v) 4 chloro-1-naphthol and 0.015% (v/v) hydrogen proxidase in PBS.
Optimizing the culture parameters for expression of P97R1R2 protein in E. coli BL21(DE3)
Optimization of cultivation temperature
To optimize the cultivation temperature for expression of recombinant P97R1R2 protein, the fresh pre-inoculum culture (2%) was inoculated in parallel shake flasks containing 20 ml LB medium with 100 µg/ml kanamycin and incubated at 37oC, 200 rpm to an OD600 = 0.8. The cultures were induced with 0.8 mM IPTG and each induced culture was incubated at different temperatures of 16, 20, 25, 30 and 37oC, 150 rpm for 8 h. The cell biomass was treated as described in our current study of expression of recombinant P97R1R2 protein and analyzed on 15% SDS-PAGE. The optimal cultivation temperature for target protein expression was used for the next optimizations.
Optimization of culture media
To determine optimal culture medium for recombinant P97R1R2 protein expression, the fresh pre-inoculum culture (2%) was inoculated in parallel shake flasks containing 20 ml of different media of LB, YJ, TB, HSG and modified M9ZB with 100 µg/ml kanamycin and incubated at 37oC, 200 rpm until OD600 reached 0.8. All cultures were induced with 0.8 mM IPTG, and expressions were made at 25oC (optimal cultivation temperature), 150 rpm for 8 h. The cell biomass was treated as described in our current study of expression of recombinant P97R1R2 protein and analyzed on 15% SDS-PAGE. The optimal culture medium for target protein expression was used for the next optimizations.
Optimization of IPTG concentration
To optimize IPTG inducer concentration for expression of recombinant P97R1R2 protein, the fresh pre-inoculum culture (2%) was inoculated in parallel shake flasks containing 20 ml of YJ medium (optimal medium) with 100 µg/ml kanamycin and incubated at 37oC, 200 rpm ultil OD600 reached 0.8. The cultures were induced with varying concentrations of IPTG of 0.2, 0.4, 0.6, 0.8 and 1 mM, and expressions were made at 25 oC, 150 rpm for 8 h. The cell biomass was treated as described in our current study of expression of recombinant P97R1R2 protein and analyzed on 15% SDS-PAGE. The optimal IPTG concentration was used for the next optimizations.
Optimization of induction time
To determine the optimal IPTG induction time for recombinant P97R1R2 protein expression, the fresh pre-inoculum culture (2%) was inoculated in parallel shake flasks containing 20 ml YJ medium with 100 µg/ml kanamycin and incubated at 37oC, 200 rpm. When OD600 of the cultures reached 0.4, 0.6, 0.8, 1.0, 1.5 and 2.0, all cultures were induced with 0.6 mM IPTG (optimal concentration) and incubated at 25oC, 150 rpm for 8 h. The cell biomass was treated as described in our current study of expression of recombinant P97R1R2 protein and analyzed on 15% SDS-PAGE. The optimal IPTG induction time for the target protein expression was used for the next optimizations.
Optimization of incubation time
To optimize post-induction incubation time for expression of recombinant P97R1R2 protein, the fresh pre-inoculum culture (2%) was inoculated in parallel shake flasks containing 20 ml YJ medium and incubated at 37oC, 200 rpm until OD600 reached 1.5. IPTG was added to a final concentration of 0.6 mM for induction and each induced culture was further incubated at 25oC, 150 rpm for 2, 4, 6 and 8 h post-induction. The cell biomass was treated as described in our current study of expression of recombinant P97R1R2 protein and analyzed on 15% SDS-PAGE.
RESULTS AND DISCUSSION
Cloning and sequencing of the P97R1R2 gene fragment
The PCR amplification of the peptide coding sequence of P97R1R2 gene fragment from genomic DNA of fresh lung tissue samples of pigs suffering from PEP has been shown in Figure 1, A. The PCR product on 1% agarose gel was a prominent band with a length of 779 bp. The purified PCR product was inserted into pGEM®-T Easy vector, and then transformed into chemically competent E. coli TOP10 cells. The presence of the insert in transformed bacterial cells was determined by PCR amplification with M13 primer pairs of the pGEM®-T Easy vector. The result of PCR showed DNA band was approximately 979 bp in length (Figure 1B), corresponding to the expected length of the insert of 779 bp and 200 bp of primer pairs on the vector. This suggested that the pGEM®-T Easy/P97R1R2 vector was successfully transformed into E. coli TOP10 cells.
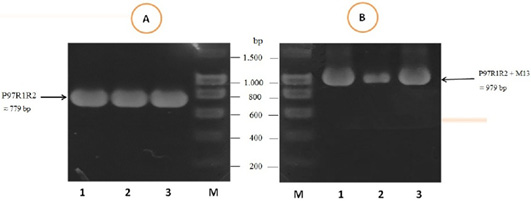
Figure 1: PCR amplification of the peptide coding sequence of P97R1R2 gene fragment (accession no. AY512905) from genomic DNA isolated from fresh lung tissue samples of pigs suffering from PEP in Thua Thien Hue province, Vietnam. M: DNA size marker (100-1500 bp, Bio Basic); Lanes 1-3 (A): PCR products of P97R1R2 gene fragment; Lanes 1-3 (B): PCR products of M13 primer pairs of pGEM®-T Easy vector.
The result of sequencing the nucleotide sequence and analysis of amino acid sequence of P97R1R2 gene fragment from M. hyopneumoniae isolated from Thua Thien Hue province, Vietnam have been shown in Figure 2. The results showed that the P97R1R2 gene fragment had 779 nucleotides, corresponding to 272 amino acids, and amino acids sequence had 10 repeats of AAKPV(E) in R1 from nucleotides 143 to 293, and 3 repeats of GS(A)PN(S)QGKKAE in R2 repeat region from nucleotides 583 to 673. Number of 10 AAKPV(E) repeats in R1 of M. hyopneumoniae P97 in the present study is similar to that found in the J strain (Hsu and Minion, 1998) and ATCC 25934 strain (Okamba et al., 2010). However, it is more than one repeat compared to the study result of Jang and Kim (2007), and less than one repeat compared to the strain 144L (Hsu and Minion, 1998). Number of 3 repeats of GS(A)PN(S)QGKKAE in R2 is similar to that found in the research result of Jang and Kim (2007). These results are in ranges of AAKPV(E) repeats in R1, and GS(A)PN(S)QGKKAE repeats in R2 of investigated M. hyopneumoniae strains P97 (Wilton et al., 1998; Jenkins et al., 2006; Jang and Kim, 2007).
Expression and purification of recombinant P97R1R2 protein in E. coli BL21(DE3)
Expression of recombinant P97R1R2 protein in E. coli BL21(DE3) cells on LB cultures with 0.8 mM IPTG induction and without IPTG induction was analysed on 15% SDS-PAGE (Figure 3). Expression of the P97R1R2 in E. coli BL21(DE3) cells is expected to produce a recombinant fusion protein with molecular weights of approximately 33.42 kDa including 29.72 kDa of P97R1R2 protein and 3.7 kDa of fusion fragment of pET-200/D-TOPO vector. On analysis of SDS-PAGE, the expressed P97R1R2 protein was not found in soluble fraction (supernatant) of transformed E. coli BL21(DE3) cells with IPTG induction (Lane 2), but found to be inclusion body form in the pellet of bacterial cell lysates (Lane 3). A prominent protein band of ~ 33.42 kDa could be seen on gel corresponding to the expected size, indicating that the 8 M urea buffer (8 M urea, 0.01 M Tris, 0.1 M NaH2PO4, pH 8.0) was efficient in solubilizing inclusion body protein in the pellet and expression of recombinant P97R1R2 protein in E coli BL21(DE3) was successful. The molecular weights of the target protein observed in this study is similar with 30 kDa protein of R1 and R2 repeats of M. hyopneumoniae P97 expressed in E. coli (Jang and Kim, 2007). For IPTG uninduced transformed E. coli BL21(DE3) cells, a very weak protein band of expected size was observed (Lane 1), suggesting that induction with IPTG is required for expression of P97R1R2 protein. The SDS-PAGE analysis also showed that purification of the recombinant P97R1R2 protein was achieved by ProBondTM Purification System Kit (Lane 4). The purified recombinant fusion protein with a size of approximately 33.42 kDa was recognized by anti-His G-HRP antibody (data not shown), confirming that the expressed protein was recombinant P97R1R2 protein.

Figure 2:Sequences of nucleotide (5’→3’) and amino acids of P97R1R2 gene fragment from M. hyopneumoniae isolated from pigs in Thua Thien Hue province, Vietnam.
Optimization of culture parameters for expression of P97R1R2 protein in E. coli BL21(DE3)
For better expression of recombinant P97R1R2 protein in E. coli BL21(DE3), the expression conditions were optimized for various culture parameters including cultivation temperature, culture media, IPTG inducer concentration, induction time and incubation time.
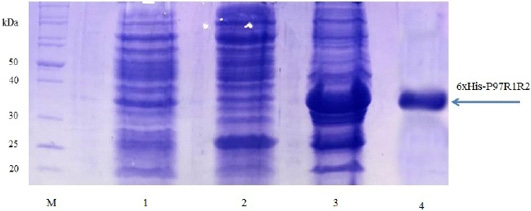
Figure 3: SDS-PAGE analysis of the expression of recombinant P97R1R2 protein in E. coli BL21(DE3) cells on LB medium. M: Protein weight marker (10-200 kDa, Bio Basic); Lane 1: transformed E. coli BL21(DE3) cells without IPTG induction; Lanes 2 and 3: Soluble (in supernatant) and inclusion body protein of transformed E. coli BL21(DE3) cells induced with 0.8 mM IPTG when OD600 of the cultures reached 0.8 and incubated at 37oC, 150 rpm for 8 h post-induction, respectively; Lane 4: purified fusion protein.
Optimization of cultivation temperature
SDS-PAGE analysis of effect of different cultivation temperatures (16, 20, 25, 30 and 37oC) on expression of recombinant P97R1R2 protein on LB medium showed that expressed P97R1R2 proteins were found to be inclusion body form at all tested cultivation temperatures (Figure 4). A previous study suggested that one way to reduce inclusion bodies was to reduce the protein translation rate by inducing expression at a lower temperature (Schein et al., 1988), however, in this study induction with IPTG at lower temperatures than 37oC (16, 20, 25 and 30 oC) also did not effect on the expression of recombinant P97R1R2 protein in soluble form. This might be due to variations in protein folding because of the differences in various characteristics such as size, hydrophobicity, post-translational modifications, isoelectric points (Donovan et al., 1996). The result of SDS-PAGE analysis in the present study also indicated that the intensity of the protein bands (level of the protein expression) increased gradually with an increase in cultivation temperatures post-induction and reached the highest level at 25oC, but thereafter there was a reduction in the expression at 30oC and 37oC. This reduction could be attributed to autolysis of bacterial cells (Pathak et al., 2008). These results agreed with a previous report that at normal temperature range, peptide chain elongation rate increases with respect to an increase in temperature which results in higher systhesis of proteins (Farewell and Neidhardt, 1998). With the obtained results, 25oC was found to be the optimal cultivation temperature for P97R1R2 protein expression, and further optimization was performed at this temperature.
Optimization of culture media
Effect of different culture media (YJ, TB, LB, HSG and modified M9ZB) on expression of recombinant P97R1R2 protein was analyzed on 15% SDS-PAGE (Figure 5). The result showed that a very weak protein band with expected size was observed (Lane NC) for the LB medium containing transformed E. coli cells without IPTG induction, confirming that induction with IPTG is necessary for expression of recombinant P97R1R2 protein. The highest expressed P97R1R2 protein was observed on YJ medium (Lane 1), and similar levels of target protein expression on the rest four culture media of TB, LB, HSG and modified M9ZB (Lanes 2-5, respectively). Therefore, YJ medium was found to be the optimal medium for expression of recombinant P97R1R2 protein. Culture media generally provide the amino acids, vitamins, minerals and trace elements to support growth and product expression in E. coli. The utilization of culture media with different compositions leads to varied cell growth rate and product yields (Donovan et al., 1996). In fact, LB mediun is one of the normal culture media used to express general recombinant proteins in E. coli (Miksch et al., 2008). However, in the pesent study, YJ medium was more suitable for expression of recombinant P97R1R2 protein in E. coli BL21(DE3).
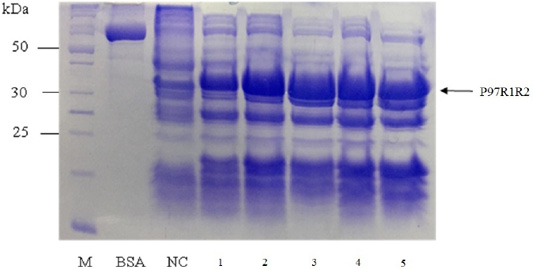
Figure 4: SDS-PAGE analysis of effect of cultivation temperatures on the expression of recombinant P97R1R2 protein in E. coli BL21(DE3) on LB medium. M: protein weight marker (10-200 kDa, Bio Basic); BSA: albumin 750 µg/ml; NC: transformed E. coli BL21(DE3) cells without IPTG induction; Lanes 1, 2, 3, 4 and 5: inclusion body protein of transformed E. coli BL21(DE3) cells induced with 0.8 mM IPTG when OD600 of the cultures reached 0.8 and incubated at different temperatures of 16, 20, 25, 30 and 37 oC, respectively for 8 h post-induction.
Optimization of IPTG concentration
Effect of IPTG induction at different final concentrations of 0.2, 0.4, 0.6, 0.8 and 1 mM at OD600 of the YJ cultures = 0.8 and optimal cultivation temperature of 25 oC for 8 h post-induction on expression of recombinant P97R1R2 protein in E. coli BL21(DE3) cells was investigated. The result showed that induction with IPTG at all concentrations produced the recombinant P97R1R2 protein (Figure 6). However, for culture induced with 0.6 mM IPTG, there was a stronger protein band at expected size (Lane 3), compared with the cultures induced with 0.2, 0.4, 0.8 and 1 mM IPTG (Lanes 1, 2, 4 and 5, respectively). These results suggest that 0.6 mM IPTG concentration is more suitable for expression of recombinant P97R1R2 protein in E. coli cells. A previous study has reported that expression of recombinant protein generally causes a metabolic burden on the cell’s energy, carbon and amino acids, which can result in reduced growth rates, product expression, and plasmid stability (Bentley et al., 1990). The level of IPTG required to adjust such metabolic burden can be vary (Donovan et al., 1996). For the pET200/D-TOPO® expression vector harboring promoter T7, final IPTG concentration should be optimized to balance the decreasing yields of recombinant cells following induction with increasing cellular levels of target protein (Bentley et al., 1991). This study found that final IPTG concentration of 0.6 mM was optimal for higher expression of recombinant P97R1R2 protein.
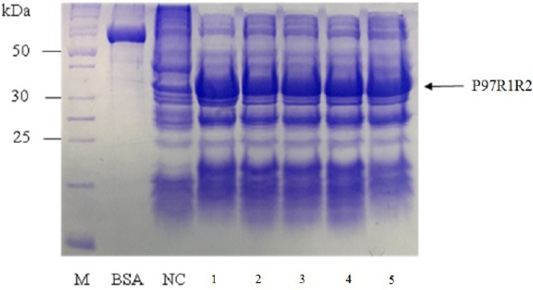
Figure 5: SDS-PAGE analysis of effect of culture media on expression of recombinant P97R1R2 protein in E. coli BL21(DE3). M: protein weight marker (10-200 kDa, Bio Basic); BSA: albumin 750 µg/ml; NC: transformed E. coli BL21(DE3) cells on LB medium without IPTG induction; Lanes 1, 2, 3, 4 and 5: inclusion body protein of transformed E. coli BL21(DE3) cells on different culture media of YJ, TB, LB, HSG and modified M9ZB, respectively, induced with 0.8 mM IPTG when OD600 of the cultures reached 0.8 and incubated at 25oC for 8 h post-induction.
Optimization of induction time
The expressed level of recombinant P97R1R2 proteins in E. coli BL21(DE3) cells on YJ medium at different induction time (OD600 of the YJ culture reached 0.4, 0.6, 0.8, 1.0, 1.5 and 2), under conditions of 0.6 mM IPTG concentration, post-induction cultivation temperature of 25oC, 150 rpm for 8 h was determined by SDS-PAGE (Figure 7). The results showed that yield of recombinant P97R1R2 protein was increased gradually with an increase of cell density (Lanes 1-4), and reached the highest yield when OD600 of culture reached value of 1.5 (Lane 5). Production of recombinant P97R1R2 protein was reduced when OD600 of culture reached value of 2.0 (Lane 6). These results are in agreement with the previous reports of Candan et al. (1998) and Malik et al. (2016) that expression level of recombinant proteins in E. coli increases up to the late exponential phase and reduced in the stationary growth phase. The maximum yield of recombinant protein from a fermentation will also depend on the point in the growth cycle at which expression is induced (Donovan et al., 1996). Results of the current study indicated that optimal IPTG induction time for the highest yield of recombinant P97R1R2 protein in E. coli BL21(DE3) on YJ medium was at OD600 of cultures reached 1.5.
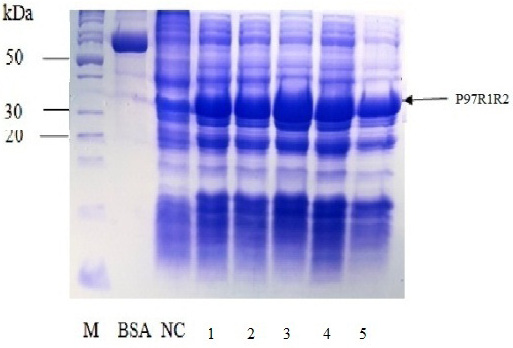
Figure 6: SDS-PAGE analysis of effect of different final IPTG concentrations on expression of recombinant P97R1R2 protein in E. coli BL21(DE3) on YJ medium. M: protein weight marker (10-200 kDa, Bio Basic); BSA: albumin 750 µg/ml; NC: transformed E. coli BL21(DE3) cells without IPTG induction; Lanes 1, 2, 3, 4 and 5: transformed E. coli BL21(DE3) cells induced with IPTG at final concentrations of 0.2, 0.4, 0.6, 0.8 and 1 mM, respectively, when OD600 of the cultures reached 0.8 and incubated at 25oC for 8 h post-induction.
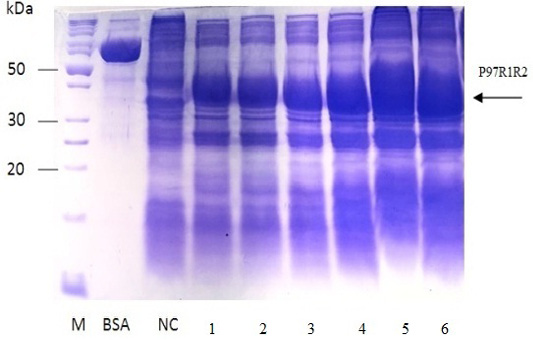
Figure 7: SDS-PAGE analysis of effect of post-induction IPTG induction time on expression of recombinant P97R1R2 protein in E. coli BL21(DE3) on YJ medium. M: protein weight marker (10-200 kDa, Bio Basic); BSA: albumin 750 µg/ml; NC: transformed E. coli BL21(DE3) cells without IPTG induction; Lanes 1, 2, 3, 4, 5 and 6: inclusion body protein of transformed E. coli BL21(DE3) cells induced with 0.6 mM IPTG when OD600 of the cultures reached 0.4, 0,6, 0.8, 1.0, 1.5 and 2.0, respectively, and incubated at 25oC for 8 h.
Optimization of incubation time
Effect of different post-induction incubation time of 2, 4, 6 and 8 h on expression of recombinant P97R1R2 protein in E. coli BL21(DE3) cells was performed under optimized culture conditions (YJ medium, OD600 of the culture = 1.5, 0.6 mM IPTG concentration and cultivation temperature of 25oC). The results were showed in Figure 8. The expression of recombinant P97R1R2 protein occured after 2 h of post-induction incubation (Lane 1), reached at the highest level after 6 h (Lane 3), and reduced after 8 h of post-induction incubation (Lane 4). Previous studies have reported that after adding the IPTG into the medium, the target protein begins to be synthesized and post-induction incubation time is necessary for recombinant protein production. The duration of post-induction incubation affects the overall folding, accumulation, and productivity of recombinant proteins in E. coli (Wong et al., 1998), and is affected by several factors such as strength of promotor, inducer concentration, solubility, and intrinsic properties of recombinant proteins (Noi and Chung, 2017). Our results suggest that the optimal post-induction incubation time for the highest expression of recombinant P97R1R2 protein in E. coli BL21(DE3) cells is 6 h.
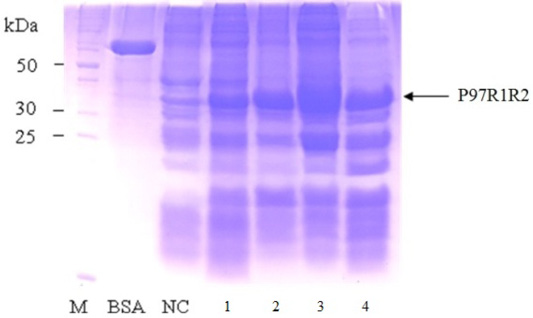
Figure 8: SDS-PAGE analysis of effect of different post-inducction incubation time on expression of recombinant P97R1R2 protein in E. coli BL21(DE3) on YJ medium. M: protein weight marker (10-200 kDa, Bio Basic); BSA: albumin 750 µg/ml; NC: transformed E. coli BL21(DE3) cells without IPTG induction; Lanes 1, 2, 3 and 4: inclusion body protein of transformed E. coli BL21(DE3) cells induced with 0.6 mM IPTG when OD600 of the cultures reached 1.5, and incubated at 25oC for 2, 4, 6 and 8 h, respectively.
CONCLUSIONS
This study was successful in cloning P97R1R2 gene fragment of M. hyopneumoniae isolated from pigs in Thua Thien Hue province, Vietnam and optimizing culture parameters (cultivation temperature, culture media, inducer concentration, induction time and incubation time) for higher expression of recombinant P97R1R2 protein in E. coli BL21 StarTM (DE3) cells to contribute to the development of new subunit vaccines against M. hyopneumoniae.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Education and Training of Vietnam (No. B2018-DHH-65).
Authors Contribution
Phung Thang Long supervised the project and edited the manuscript. Le Quoc Viet, Le Viet Quan, Dong Huu Rin and Nguyen Thi Thu Hien performed the experiments and interpreted the data. Nguyen Xuan Hoa, Le Duc Thao and Le Dinh Phung provided guidance and support to carry out the experiments. Dinh Thi Bich Lan designed the experiments and wrote the draft of manuscript.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest associated with this study.
REFERENCES



