Advances in Pharmaceutical and Ethnomedicines
Review Article

Genetic Bone Deformities and its Treatment at Molecular Level
Shumaila Arshad1*, Aqsa Ashiq2
1Faculty of Pharmacy, The University of Lahore, Lahore, Pakistan; 2Institute of Molecular Biology & Biotechnology, The University of Lahore, Lahore, Pakistan.
Abstract | Bone is formed by minerals, predominantly specialized proteins, calcium hydroxyapatite which make the bone matrix for proper function and bones strengthen. Bone is composed from osteoclasts, osteoblasts and collagen. Vessels are fundamental for basic bone multicellular units which organizes formation of osteoblasts and osteoclasts. Bone resorption is responded by osteoclasts (polynuclear cells). Osteoclasts function is vital in the maintenance, repair and bone remodeling.Imbalance in bone matrix leads towards disorders of bone such as osteogenesis imperfecta, paget’s disease of bone, osteoporosis, diastrophic dysplasia, cleidocranial dysplasia, kniest dysplasia, pycnodysostosis, caffey disease and achondroplasia which leads toward severe and sometime skeleton anomalies, back bone pain, bone fracture, extra toe, bent of tibia, head and neck anomalies, osteogenesis of skeleton muscles, stature, deteriorating joints, pelvic deformities, skull, craniofacial defects, shortening and extensiveness in bones, brachydactyly, osteoarthritis, clubbed foot, carpal and tarsal osteolysis and muscle weakness. Bone deformities occur at genetic level with different mode of inheritance pattern. Changes at genetic level can cure only by gene therapy by using in vivo or ex vitro gene therapy methods using either by viral or non-viral vectors, most common used is adeno-associated virus. Bone anomalies are one of the major problems in Pakistan and it is increasing rapidly day by day. Approximately 72% Pakistanis are suffering from bone anomalies, mostly women are affected. Paget’s disease of bone, osteogenesis imperfecta, spondyloepimetaphyseal dysplasia and many other disorders are reported in Pakistan. The high prevalence of genetic bone anomalies in Pakistan is due to lack of awareness and cousion marriage.
Keywords | Bone, Bone Disorders, Gene therapy, Bone anomalies in Pakistan, Treatment
Editor | Gulzeb Aziz, Norwegian Medicinal Agency, Oslo, Norway.
Received | June 16, 2016; Accepted | August 02, 2016; Published | August 20, 2016
*Correspondence | Shumaila Arshad, Faculty of Pharmacy, The University of Lahore, Lahore, Pakistan; Email: shumailapharmacist@gmail.com
Citation | Arshad S and Ashiq A (2016). Genetic Bone Deformities and its Treatment at Molecular Level. Adv. Pharm. Ethnomed. 3(1): 6-18.
DOI | http://dx.doi.org/10.14737/journal.ape/2016/3.1.6.18
ISSN | 2310-0575
Copyright © 2016 Arshad and Ashiq. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
A disease is a particular abnormal condition, a disorder of a structure or function that affects part or all of an organism.Disease is a condition of disturbance of physical or mental status which reduces functional abilities of body. Diseases include disabilities, syndromes, disorders, injuries or infections. These diseases can cause pain, distress or social problems and can even lead to death of the patient. A disease can occur due to genetic or environmental changes. Environmental diseases are caused by biotic or abiotic factors. Abiotic factors include soil, polluted air, poor sanitary and traffic pollution. All these factors have great impact on health and are unpredictable. Genetic diseases are inherited from parents to offspring. A genetic disease occurs due to mutation in respective gene or due to chromosomal anomalies. Genetic diseases are passed on by different inheritance pattern e.g., recessive, dominant, X-linked, autosomal or by mitochondrial inheritance. A single mutation has many consequences. Mutation can be harmful, beneficial or neutral. Harmful mutations cause many disorders e.g. duchene muscular dystrophy (DMD), cancer, down syndrome and different types of bone abnormalities. Genetic diseases cannot be cured with medicine and surgery they can be treated only by gene therapy.
Bones are composed of bone matrix. Bone matrix is essentially comprised of fibers, organic and inorganic components, specialized proteins, mineral, hydroxyapatite and collagen (Cumming et al., 2002). Bone strength is maintained by composition of the contents. Bone coordination is fundamental for effective circular system and vascular functions. There are four categories of bones: irregular bones (sacrum, hyoid, coccyx and vertebra), long bones (clavicles ulnae, femur, humeri, metatarsalss, radii, metacarpals, phalanges and fibulae), flat bones (ribs, skull, mandible, sternum and scapulae) and short bones (sesamoid, tarsal, patellae and carpal). Skeleton system is responsible to perform numerous functions e.g., acid-base and mineral homeostasis balance. Skeleton system is made up of 20% trabecular bones and 80% cortical bones (Clarke, 2008). Bones are essential for life to perform numerous functions, such as support, tendons, ligament, muscles attachments, protection, support of brain, spinal cord and heart, maintenance of hematopoiesis, minerals and particularly regulation of calcium and restoration of fractures (Cohen, 2006). The properties of bones change throughout the life (either weakens or further improving functions) (Boskey and Coleman, 2010). Vessels are basic fundamentals for bone multicellular units, which organize formation of osteoblasts and osteoclasts. Consequently, for appropriate functioning, vascular system of bone is essential for strong bone and bone marrow. Bone resorption is responded by osteoclasts (polynuclear cells). Osteoclasts function is vital in the maintenance, repair and bone remodeling. Modifications in skeleton are linked with fluctuations in cell metabolism of bone. A bone fracture is the outcome of minor injuries. Fragility in bones is the consequence of variations in the structural properties and material of the bone. Imbalance in bone matrix leads towards disorders of bone such as osteogenesis imperfecta (OI), Paget’s disease of bone (PDB), osteoporosis, diastrophic dysplasia, cleidocranial dysplasia, kniest dysplasia, pycnodysostosis, caffey disease and achondroplasia. These anomalies are outcome of dense bones, failure of osteoclast function, weakness of bones and improper bone matrix composition. The nature of bone potential is determined by bone strength. Osteopetrosis potentially impairs bone marrow function. When osteoblast production is reduced, bone mass is reduced. Osteoporosis consequences in potential susceptible to bone fractures (Cohen, 2006). Osteoporosis effects growth of skeleton. Hormonal imbalance, mechanical factors and vascular anomalies play role in it (Atanda et al., 2011).
Mutated COLA2 gene can affect from perinatal lethal to mild joint anomalies. Stickler syndrome results from nonsense mutations in COLA1 and COAL2 genes. Mutation in aggrecan results in autosomal recessive disorders of severe short stature facial anomalies. Spondyloepimetaphyseal dysplasia (SEMD) results from missense mutation. Kniest dysplasia results in mutation in amino terminal end.
Osteoporosis is a foremost health issue, especially in elders. It is assumed by the half of 2050 fractures of hips will commonly occur in Asia. Higher hip fractures are described from industrialized countries. The highest rates of hip fractures are in North Europe and the US while low rates are in Latin, America and Africa. Asian countries such as Kuwait, Iran, China, and Hong Kong show less hip fracture rates as compared to these countries. There is also a North–South gradient seen in European studies and more fractures are seen in the North of the US than in the South. Hip fractures in US populations are premier in the world. Researchers reported that age-standardized annual incidence of hip fracture per 100,000 are 197-201 in men and 511-553 in women respectively. In Pakistan, orthopedic disorders are becoming alarming as 72% population suffering from bone disorders out of which 55% are women.
Problems
How deformities in skeleton, long bones and bone fragility leads toward anomalies of bones. What are the inheritance patterns and how genes affect the normal functions of bones? Which genetic factors or mutations are responsible of it? The treatment of bone disorders is very costly and its prevalence is increasing day by day. The risks of fractures are 1.5 million every year. Ten million individuals over the age of 50 in United States have osteoporosis of hip while 33.6 million individuals above 50 years have osteopenia. By 2020, one out of two Americans above 50 years probably has high risks of osteoporosis. In Pakistan cousin marriages are responsible for it. To solve these issues there are following objectives:
GENETIC BONE ANOMALIES
Skeleton anomalies belong to heterogeneous group. Genetic bone anomalies can occur by autosomal dominant, autosomal recessive, X-linked dominant and X-linked recessive mode of inheritance. Genetically bones or skeleton anomalies also belong to uniparental disomy, chromosomal duplication, chromosomal deletion, germline mosaicism and interfamilial or intrafamilial variability. Patients with lethal or severe bone anomaly indicates their parents have two different dominant mutation either autosomal dominant or X-linked dominant. After the completion of human genome project, loci of several bone and cartilage were identified. Hereditary mutations which affects cartilage growth develops in several skeleton disabilities because cartilage is pivotal for postnatal skeleton growth. Collagen occurs frequently in body. Mutation in collagen genes (COL1A1 and COLA2) is mostly responsible for genetic bone anomalies (Azouz et al., 1998). Genetic anomalies of the skeleton are disparate group of disorders whose unifying features are malformation, inconsistent growth, and distortion skeleton deformed bones or of individual bones or groups of bones. Collagen gene mutations have been found to be responsible for osteogenesis imperfecta and many other bone anomalies (Azouz et al., 1998). Null mutation in aggrecan (proteoglycan) leads toward heterozygosityin spondyloepipyseal dysplasia. Missense mutation in FLNA (Filamin B protein) gene is responsible for many bone anomalies (boomerang dysplasia, frontometaphyseal dysplasia and otopalatodigital syndrome). For many skeleton anomalies mutation in FLNB is responsible (Krakow et al., 2004) while some other genetic bone anomalies are as follow:
Osteogenesis Imperfecta
Osteogenesis Imperfecta (OI) is one of the most common genetic bones anomaly. Inheritance pattern of OI is autosomal dominant as well as autosomal recessive. Maximum types of OI are linked to collagen that is a rod like structure. About 90% mutations for OI were reported in COLA1 and COLA2 genes while other genes responsible for OI are IFITM5, SERPINF1, CRTAP, LEPRE, PPIB, SERPINH1, FKBP10, SP7, BMP1 TMEM38B, WNT1, CREB3L1, PLS3 and PLOD2. Prevalence of OI is 1 in 10,000-20,000 live births (Atta at el., 2014). OI is clinically characterized by fragile bone, skeleton fragility, scoliosis, hearing loss, joint sloppiness, hypercalciuria, neurological problems, tibia bent basilar invagination macrocephaly, femur bent anomalies in spine, osteoporosis, skin ligaments and dentogenesis imperfecta (Atta et al., 2014).
The severity in different types of OI is different which generally subjected fragility of bone. Type I of OI is less sever, OI type II is perinatal lethal, OI type III is sever and OI type IV is moderate whereas type V is not severe. There is not too difference between type II and type VII (except white and blue sclera and small head), type III and type VIII is approximately clinically similar (round face and shape of chest). Type IX is less severe as compared to type VIII. The clinically severity of OI types can be distinguished as type I < types IV, V, VI< III, VII, VIII< type. During second trimester of pregnancy prenatal identification for lethal OI in long bone fractures can be done via ultrasound examination. The prenatal analysis of OI can be carried out by radiography and biochemical studies of cultivated fibroblasts from the fetus (Berge et al., 1995).
Paget’s Disease of Bone
Paget’s disease of Bone (PDB) is disorganized bone remodeling anomaly. It has numerous susceptible loci for the disorder, identified by genome-wide scans.
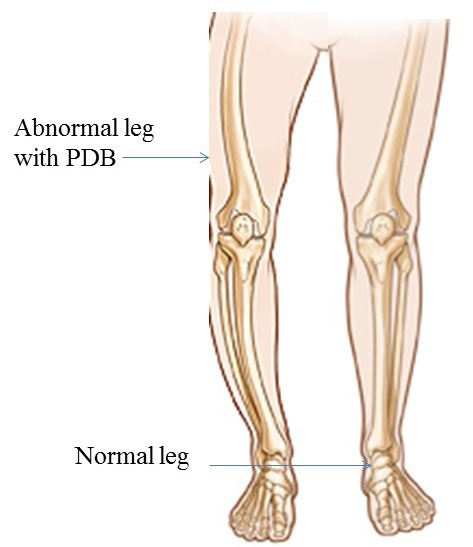
Environmental dynamics also play vital role in PDB etiology. Patient with PDB has reduced quality of life. Chromosomal locations are 18q21.2-21 (TNFRSF11), 8q24 (TNFRSF11B) (Cundy et al., 2002), 5q35.5 (SQSTM1) (Layfield and Hocking, 2004) and 9p13.3 (VCP), with mode of inheritance autosomal dominat. Four causative genes TNFRSF11A, TNFRSF11B, SQSTM1 and VCP are identified. These genes play a critical and important role in the bone metabolism and bone diseases. Prevalence of PDB is reported as 5/10,000 (men) 3/10,000 (women) (Cooper et al., 2006). Clinical signs and symptoms of PDB are backbone osteoarthritis (hips and knee) (Altman, 1980), bone bending and fracture (Altman,1999), long bones, polysomic, osteosarcoma none-skeleton, 12 pairs of nerve, peripheral nerve (motor and sensory nerve) nerve root, congestive heart failure, hypercalcemia and hearing loss. The prenatal diagnosis for PDB is carried out by ultrasound, laboratory studies of markers show increased bone turnover (total volume of bone) while radiography shows thickening of the skull (Figure 1).
Diastrophic Dysplasia
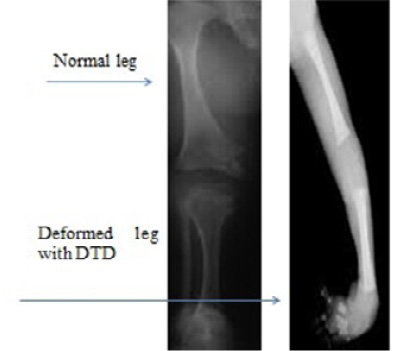
Diastropihic Dysplasia (DTD) is characterized by extensive long bone, skeletal abnormalities, hypognathia, cervical kyphosis, persistent extensive drawback in elbow and knee junctions, club foot, deviation of ulna hands, small phalanges, backward bent of thumb and toes, skull and spinal abnormalities, spinal curvature anomaly, hyperbolic and contraction of the extensive joints abnormalities, osteoarthritis, ulnar deviate fingers, distance between first and second toes and clubfoot (Bonafe et al., 1993). Inheritance pattern of DTD is autosomal recessive, with cytogenetic location 5q32. Causative gene is SLC26A2 (Bonafé et al., 2008) while prevalence of DTD is reported as 1 in 100,000 live births. Prenatal diagnosis is based on 31 weeks of gestation by sonographic ultrasound and DNA test by molecular analysis (PCR) (Figure 2).
Cleidocranial Dysplasia
Cleidocranial Dysplasia (CCD) is characterized by irregular clavicles, permit stature and fontanelle, excessive teeth, dwarf stature, effects of temporal bone, diversity of skeletal changes and flaws. Inheritance pattern of CCD is autosomal dominant. Cytogenetic location of gene is 6p21 while prevalence is reported as 1/1,000,000. So far molecular analysis for RUNX2 gene is not informative. The available prenatal diagnosis is three dimensional ultrasonogrphy examination during pregnancy which can confirmed CCD. Three dimensionalultra sound have sound results for hysterosalpingography and for major congenital anomalies which facilitates analysis of uterine abnormalities, permitted easy differentiation in subseptate (uterus divided by fibrous or muscular wall) and bicornuate uteri (uterus is heart shape instead of pear shape). Cleidocranial dysplasia is a condition that primarily affects the development of the bones and teeth. Signs and symptoms of cleidocranial dysplasia can vary widely in severity, even within the same family.
Individuals with cleidocranial dysplasia usually have underdeveloped or absent collarbones (clavicles). As a result, their shoulders are narrow and sloping, can be brought unusually close together in front of the body, and in some cases the shoulders can be made to meet in the middle of the body. Delayed closing of the spaces between the bones of the skull (fontanels) is also characteristic of this condition. The fontanels usually close in early childhood, but may remain open into adulthood in people with this disorder.
Affected individuals may be 3 to 6 inches shorter than other members of their family, and may have short, tapered fingers and broad thumbs; short forearms; flat feet; knock knees; and an abnormal curvature of the spine (scoliosis). Characteristic facial features may include a wide, short skull (brachycephaly); a prominent forehead; wide-set eyes (hypertelorism); a flat nose; and a small upper jaw.
Individuals with cleidocranial dysplasia may have decreased bone density (osteopenia) and may develop osteoporosis, a condition that makes bones progressively more brittle and prone to fracture, at a relatively early age. Women with cleidocranial dysplasia have an increased risk of requiring a cesarean section when delivering a baby, due to a narrow pelvis preventing passage of the infant’s head.
Dental abnormalities seen in cleidocranial dysplasia may include delayed loss of the primary (baby) teeth; delayed appearance of the secondary (adult) teeth; unusually shaped, peg-like teeth; misalignment of the teeth and jaws (malocclusion) and extra teeth, sometimes accompanied by cysts in the gums (Figure 3).
Hereditary Multiple Exostosis
Hereditary Multiple Exostosis (HME) is characterized as dwarf stature, limb-length divergences, valgus distortions of the ankle and knee, irregularity of the pectoral and pelvic girdles, bowing of radius, ulnar deviation, wrist and subluxation of the radiocapitellar joint. Inheritance pattern of HME is AD (Alvarez et al., 2007). Causative genes of HME are EXT1 and EXT2 (Alvarez et al., 2007). Chromosomal locations are 8q24 (EXT1) and 11p11-13 (EXT2). Prevalence of HME is 1 in 100,000. Previously, for prenatal diagnosis the gene was amplified via PCR then direct sequencing was carried out. After doing genotypingof proband, amniocentesis sampling was performed for prenatal genetic diagnosis (Figure 4) (Table 1, given at the end.
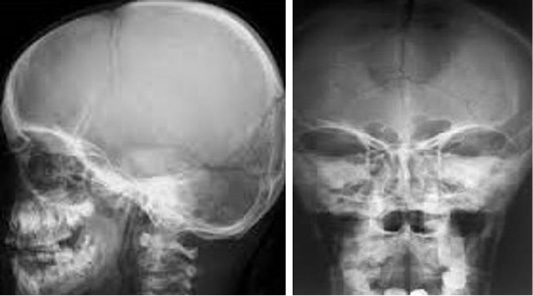
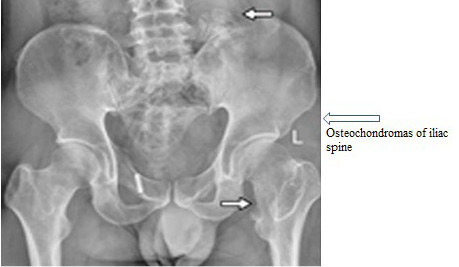
Figure 4: Shows abnormal pelvic with HME
Gene Therapy
Basics of Gene Therapy
Gene therapy is a practical method to insert a small fragment of RNA or DNA sequence in place of mutated sequence to treat genetic anomalies. At present, the majority of gene therapeutic techniques are focusing on inserting genetic material in cells instead of changing in genome. Gene therapy is an effective treatment option for genetic diseases which have no other treatment and cure.Introduce a small fragment of DNA or RNA into the effected cells of an individual with the intention of producing a therapeutic benefit for the patient (Dai and Rabie, 2007). Prospective use of gene therapy to cure diversity of orthopedic disorders is currently an active area of investigation.
Types of Gene Therapy
Gene therapy is classified into two categories: germline gene therapy and somatic gene therapy. For the cure of genetic disorder gene therapy is carried out either by ex vivo or in vivo technique which are explain in following
Somatic gene therapy: Somatic gene therapy (SGT) (Figure 5)is the type of gene therapy, used for treating cells except sperm and egg in the affected body. The proportion of transgenic cells ameliorates the complete disease signs. For example the recombinant adeno-associated virus (rAAV) is effectual to cure bone anomalies with relevance ofin vivo targeting of somatic tissue.
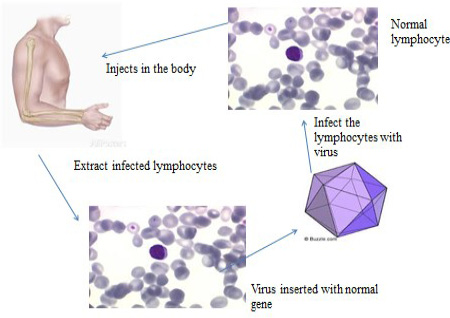
Germline gene therapy: Germline gene therapy (Figure 6) technique is used to introduce transgenic cells in germ cells e.g. sperms and eggs.This therapy is not only to treat a patient however gametes also fix the changed genotype.Germline gene therapy introduce genes in reproductive cells (egg and sperm) or in the future into embryo to correcting genetic anomalies that can be passed on to future generations. Thus till now no human germline gene therapy is performed but animal models are in practice.
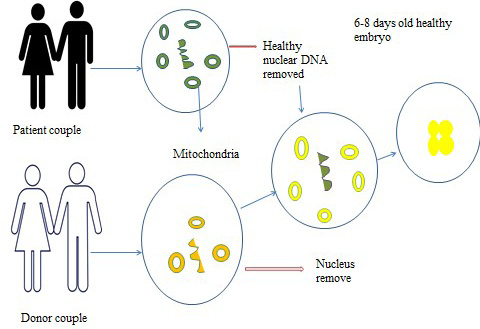
Figure 6: Germline gene therapy
Gene Therapy for Genetic Bone Anomalies
Genetic disorders can’t be cure by medicines. They can be treated only by gene therapy. Following are the ongoing molecular based research and treatment plans for genetic bone anomalies with the help of gene therapy.
Gene therapy for mutated SLC26A2 gene: There are no therapeutic approaches for treatment of DTD but some mouse models are in process.
Gene therapy for mutated ACH: Adeno associated virus (AAV) is used for ACH gene therapy and it is maximum effectual at the age of 1-5 years. In mice FGFR3 gene leads towards significantincrease in long bones and vertebral column.
Gene therapy for mutated HPP: A mimic mice with HPP which dies after two weeks of birth. Adeno-associated virus serotype 9 (AAV9) was injected in the uterus of mice. After prenatal diagnosis of HPP it is concluded that it is an effective for lethal treatment HPP.
Gene therapy for mutated metatropic dysplasia and FOP: No treatment for gene therapy has been done for metatropic dysplasia and FOP, (https://rarediseases.org/rare-diseases/fibrodysplasia-ossificans-progressiva/accesed).
Gene therapy for mutated LMD: The result for growth hormone therapy was seen with combined turner syndrome (TS) and LMD. Karyotype testing results showed 45,X/46,X,idic(X). Genetic analysis for SHOX gene did not show any exonic mutation. A marker downstream of the 3’ end of SHOX gene coding sequence, were not present with resultant LMD. There is no improvement for skeleton deformities. It is concluded that growth hormone therapy is not advantageous in severe short stature because of combined turner syndrome (TS) and LMD.
Stem cell gene therapy through AAV for OI: Adeno-associated virus belongs to parvovirus family. It is the smallest non-enveloped, linear single stranded DNA. The shape of AAV viron shape is icosahedral and 20-25nm in diameter. The host of AAV-1 is not clear but AAV-2 and 3 are isolated from specimens of humanoid clinically. Adeno-associated virus-4 is isolated from kidney cell culture (Dai and Rabie, 2007).
The best approach for OI is the combination of stem cells with gene therapy. Mesenchymal Stem Cell (MSCs) have capacity to differentiation into adipocytes, osteocytes, myocytes, nerve cells and tenocytes in vitro. Bone morphogenetic protein may lead to engraftment of functional MSC differentiate into bone cells in bone marrow and MSCs have potential to self-renewal. For cellular activities of skeletal tissue regeneration MSCs are very effective. Earlier reports demonstrated the difficulties and complications in transducing effectiveness of rAAV into MSC. Adeno-associated virus is used to dislocate the exon 1 of COL1A1 gene in MSC which is isolated from patients with OI whichdemonstrate its effectiveness in gene targeting for adult humanoid stem cells (Dai and Rabie, 2007). Redifferentiation and dedifferentiation of somatic cells, BMT and MSC are the approaches for OI.
Gene therapy for bone and joint anomalies: Bone allograft is usually used in orthopaedic surgeriesso for this purpose AAV is able of transduced inflammatory cells and osteoblasts. Bone morphogenetic protein (BMT) signals coating through AAV-caALK2 (constitutively active activin receptor like kinase-2), induces directly bone formation on cortical surfaces of allograft. In vivo allografts coated with AAV-caALK2 are mediated.Current research investigates early stages on the way to improve the gene therapy cure opportunities for problematic orthopedic disorders (Figure 7).
Recombinant adeno-associated virus (rAAV) for gene therapy of bone: Recombinant adeno-associated virus has favorable therapeutic approaches for treatment of bone and cartilage problems with possible advantages of target gene therapy. Adeno associated virus-bone morphogenetic protein-2 (AAV-BMP-2) vector is directly injected in hind limb of rat muscle in vivo. Remarkable heterotopic novel bone development is detected and BMP-2 expression is persisted. Adeno associated virus-bone morphogenetic protein-2 (AAV-BMP-4) can effectively increase endochondral ossification development instantly when vector is injected in sprague dawley (SD) rats. Adeno associated virus is an effectual, safe and easy to introduce in skeleton and has benefits above viral and non-viral vectors (Dai and Rabie, 2007).
STATUS OF GENETIC BONE ANOMa-LIES IN PAKISTAN
The population of Pakistan was 191.71 million in 2015. Day by day the population of Pakistan is increasing rapidly. Increase in population causing several problems. Cancer and phenylketonuria are common in Pakistanis but now osteoporosis and arthritis are also common diseases. Bone disorders are one of the serious problems in Pakistan. Orthopedic disorders are becoming common as 72%, population is suffering from bone disorders out of which 55% are women. Osteopenia and osteoporosis are problematic. Osteoporosis is highly prevalent as 42% women are suffering from Osteopenia and 29% women are suffering from osteoporosis. Osteopenia is highly prevalent in young women. A survey in which 334 women over age of 20 years, it is revealed that out of 334, 43.3% women have osteopenia and 12.9% are suffering from osteoporotic. Sixtty four % women of less than 30 years, 55% women between 31-45
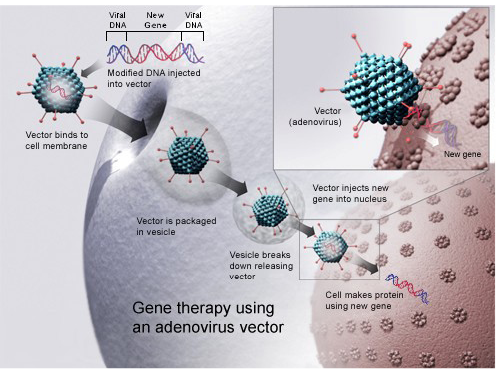
Figure 7: Gene therapy using an adenovirus vector
years and 73.9% women over age of 45 years found with low BMP. This study revealed that women have high risk of bone problems. Prevalence of vertebral, hip fractures and bone fragility is unknown. Men up to 60 years and women up to 45 years are facing bone and joints problems. A 70 years old patient is reported with PDB, with pain in right leg and problem in walking, this situation was since 5 months. His whole body was scaned which revealed the signs and symptoms of PDB. Further diagnosis is confirmed by bone biopsy. It is revealed in Pakistan PDB is present. Extrapolate prevalence of PDB in Pakistan is 4,775,890. A few cases (140) with DTD were reported in Finland but more DTD cases (1,591) are reported in Pakistan. A patient from Abbottabad, with multiple medical issues is reported in Ayub Medical Complex (AMC), after detailed medical observations it is found that she is suffering from DTD. The rest of her family members are may be the carrier while one of her sister was died with DTD. Spondyloepimetaphyseal dysplasia (SEMD) is reported with AR inheritance pattern, causative gene is PAPSS2 with cytogenetic location 10q22-q24. Two families with hereditary multiple exostosis were reported and the reason is consanguineous marriages. They have the symptoms of HME. Cleidocranial dysplasia’s first icase was reported in 2005. In Pakistan no native statistics are available on effectiveness of OI therapy for children (Atta et al., 2014).
Prevelanceof Genetic Bone Anomalies
The prevalence of genetic bone anomalies is unknown due to lack of tools, disease registry centres and unawareness of people. People believe that it is as an aging effect.Extrapolate of PDB and DTD is 4,775,890 and 1,591, respectively.
Major Inititutes Dealing with Bone anomalies in Pakistan
Many institutes are established in Pakistan which are working for the treatment of genetic bone anomalies, some of which are: Pakistan Society for the Rehabilitation of the Disabled (PSRD, Combined Military Hospital (CMH), Dow University of health sciences, Pakistan Orthopedic Association (POA), Pakistan Society of Rheumatology (PSR) , Ayub Medical Complex (AMC), Chandka Medical College of Shaheed Mohtarma Benazir Bhutoo Medical University Larkana , National Institute of Child Health, Lahore General Hospital (LGH) and Ameer Uddin Medical College.
Clinical Diagnosis of Bone Anomalies in Pakistan
Genetic bone anomalies can be diagnosed by radiographic. Dual-energy X-ray absorptiometry (DXA), Fracture Risk Assessment Tool (FRAX) and genetic test
Gene Therapy in Pakistan
No work has done on gene therapy for genetic bone anomalies in Pakistan.
Acknowledgment
It would not have been possible without the blessings of Allah almighty & His kindness. & for the next we all are indebted to acknowledge Hazrat Muhammad (saw) whose teachings and blessings enlighten our souls through hardships, & in every moment of our life. & our respected Mam Shumaila Arshad, it was her encouragement, & faith in us that we did this small & humble effort. & our all cheerful friends who helped us during this effort, & Our Parents love & prayers for all of us.
Conflict of interests
There is no conflict of interest.
Authors’ contribution
All the authors contributed equally.
References
Table 1: Genetic bone abnormalities with their pattern, causative gene, prevalence, cytogenetic locatiion and clinical signs and symptoms
|
Sr.# |
Name of the disease |
Inheritence pattern |
Gene |
Cytogenetic location |
Prevalence |
Clinical signs and symptoms |
|
1 |
Paget’s disease of bone (PDB) |
AD (Ralston and Albagha, 2014). |
TNFRSF11A, TNFRSF11B, SQSTM1and VCP (Daroszewska and Ralston, 2006). |
18q21.2-21.3(TNFRSF11A) (Hughes et al., 2000), 8q24 (TNFRSF11B)(Cundy et al., 2002), 5q35.3 (SQSTM1) (Layfielda andHocking, 2004), and 9p13.3 (VCP) (Kovach et al., 2001). |
5 /10,000 (men) 3/10,000 (women) (Cooper et al., 2006). |
Backbone osteoarthritis (hips and knee) (Altman, 1980), bone bending and fracture (Altman, 1999), elongated bone (Hughes etal., 2000), polysomic, osteosarcoma none-skeleton, 12 pairs of nerve, peripheral nerve (motor and sensory nerve) nerve root, congestive heart failure, hypercalcemia, hear loss. |
|
2 |
Hypophosphatasia (HPP) |
AD and AR (Mornet E, 2007). |
ALPL (Mornet E, 2000). |
1p36.12 (OMIM# 241510). |
1/100 000(Morne E, 2007). |
Early deciduous loss of teeth (Van den Bos et al., 2005), loss of bone and teeth mineralization(Brun-Heath et al., 2007). |
|
3 |
Boomerang dysplasia (BD) |
AD (Robertson, 1993). |
FLNB (Robertson, 1993). |
3p14.3 (OMIM# 2310). |
Unknown |
Osteogenesis of bone limb and spinal column harmful skeletal dysplasia. Higgledy-piggledy skeletal, under development of acetabulum, and incomplete development of tissues and organs(Bicknell et al., 2005). |
|
4 |
Acromicric dysplasia (AD) |
AD (Klein et al., 2014). . |
FBN1 (Klein et al., 2014). |
15q21.1 (OMIM# 102370). |
22 cases(Faivre et al., 2001). |
Small stature, small extremities, inflexible joint brachymetacarpia, coned epiphyses, internally nick of the femoral skull, late bone age(Klein et al., 2014). |
|
5 |
Diastropihic dysplasia (DTD) |
AR (Montiet al., 2015). |
SLC26A2 (Bonafé et al., 2008). |
5q32 (Bonafé et al., 2103). |
1/100,000 live births (Honorio et al., 2013). |
Extensive long bone, skeletal abnormalities, hypognathia, cervical anatomy, persistent extensive drawback in elbow and knee junctions, club foot, deviation of ulna hands, small phalanges, backward bent of thumb and toes (Gembruch et al., 1988), skull and spinal abnormalities, spinal curvature, hyperbolic and contraction of the extensive joints abnormalities, osteoarthritis ulnar deviate fingers, distance between first and second toes, clubfoot (Bonafe et al., 1993). |
|
6 |
Cleidocranial dysplasia (CCD) |
AD (Lo Muzio et al., 2007). |
CBFA1/RUNX2 (Lo Muzio et al., 2007). |
6p21 (Lo Muzio et al., 2007). |
1: 1,000,000 (Garg and Agrawal, 2008). |
Irregular clavicles, permit suture and fontanelle, excessive teeth, dwarf stature, diversity of skeletal changes and flaws (Lo Muzio et al., 2007),effects of temporal bone (Segal and Puterman, 2007). |
|
7 |
X-linked chondrodysplasia punctata 1 (CDP1) |
X-linked recessive (Franco et al., 1995). |
ARSE (Franco et al., 1995). |
Xp22.3 (Franco et al., 1995). |
Unknown |
Deformities in bone and cartilage development (Brunetti-Pierri et al., 2003), speckled epiphyses underdeveloped of midface and of the nasal bones, small stature, brachytelephalangy, ectopic calcifications(Casarin et al., 2009). |
|
8 |
Osteogenesis imperfecta type I (OII) |
AD (Cohen, 2006). |
COL1A1 (Cohen, 2006). |
17q21.33 (OMIM# 166200). |
1/120,000-1/30,000 (Cohen, 2006). |
Blue sclera, normal teeth, anomalies of stature, easily damaged skin, mild joint hypermobility and kyphoscoliosis, hernias problem, and arcus senilis, aortic valvular insufficiency mitral valve prolapse (Hortop et al., 1986). |
|
9 |
Osteogenesis imperfecta type II (OI II) |
AD (Cohen, 2006). |
COL1A1/ COLA2 (Cohen, 2006). |
7q21.3 17q21.33 (OMIM# 166210). |
1/60,000 (Cohen, 2006). |
A thin-boned and a broad-boned type(Remigio and Grinvalsky, 1970). |
|
10 |
Osteogenesis imperfecta type III (OI III) |
AD and AR (Cohen, 2006). |
COL1A1/ COLA21/ (Cohen, 2006). |
7q21.3 1721.33 (OMIM# 259420). |
1/70,000 (Cohen, 2006). |
Blue sclerae, deformity of the limbs in childhood and of the spine in late childhood and adolescence, dentinogenesis imperfecta (Sillence el al., 1979). |
|
11 |
Osteogenesis imperfecta type IV (IV) |
AD (Cohen, 2006). |
COL1A1/ COLA2 (Cohen, 2006). |
7q21.3 17q21.33 (OMIM# 166220). |
Unknown |
Shortening of the limbs with severe angular malformations, pale blue sclerae, lumbar spondylolisthesis, and dentinogenesis imperfecta (Johnson et al., 2002). |
|
12 |
Osteogenesis imperfecta type V (OIV) |
AD (Cohen, 2006). |
IFITM5 (Shaker et al., 2015). |
11p15.5 (OMIM# 610967). |
Unknown |
Severe increased fragility of long bones and vertebral bodies (Glorieux et al., 2000). |
|
13 |
Osteogenesis imperfecta type VI (OIVI) |
AD or AR (Cohen, 2006). |
SERPINF1 (Shaker et al., 2015). |
(OMIM# 613982). |
Unknown |
Frequent fractures, dentogenesis imperfecta was uniformly absent, vertebral compression fractures, moderate to severe form of OI (Glorieux et al., 2002). |
|
14 |
Osteogenesis imperfecta VII (OIVII) |
AR (Cohen, 2006). |
CRTAP (Shaker et al., 2015). |
3p22.3 (OMIM# 610682). |
Unknown |
Moderate and severe, bone fracture, bone deformity, stature anomalies (Robert, 2013). |
|
15 |
Osteogenesis imperfecta VIII (OIVIII) |
AR (Shaker et al., 2015). |
LEPRE1 (Shaker et al., 2015). |
1p34.2 (OMIM# 610915). |
Unknown |
Stature abnormalities multiple bone fractures (Robert, 2013). |
|
16 |
Osteogenesis imperfecta type IX(OIIX) |
AR (Shaker et al., 2015). |
PPIB (Van Dijk et al., 2009). |
15q22.31 (OMIM* 123841). |
Unknown |
Bone fragility, fractures (Van Dijk et al., 2009). |
|
17 |
Osteogenesis imperfecta type X(OIX) |
AR (Shaker et al., 2015). |
SERPINH1 (Shaker et al., 2015). |
11q13.5 (OMIM# 613848). |
Unknown |
Bone fragility and low bone mass, multiple bone anomalies, fractures, osteopenia, dentinogenesis imperfecta, and blue eye sclera (Christiansen et al., 2010). |
|
18 |
Osteogenesis imperfecta type XI (OIXI) |
AR (Shaker et al., 2015). |
FKBP10 |
17q21.2 (OMIM *607063). |
Unknown |
Fragility of bones, low bone mass, and connective-tissue indication (Rauch and Glorieux, 2004). |
|
19 |
Osteogenesis imperfecta type XII (OIXII) |
AR (Shaker et al., 2015). |
SP7 (Shaker et al., 2015). |
12q13.13 (OMIM# 613849). |
Unknown |
Slightly bone anomalies, delayed teeth eruption, no dentinogenesis imperfecta and white eye sclerae (Lapunzina et al., 2010). |
|
20 |
Osteogenesis imperfecta type XIII (OIXIII) |
AR (Shaker et al., 2015). |
BMP1 (Shaker et al., 2015). |
8p21.3 (OMIM# 614856). |
Unknown |
Bone fragility and low bone mass, blue sclerae, severe growth deficiency, borderline osteoporosis (Martinez-Glez et al., 2012). |
|
21 |
Osteogenesis imperfecta type XIV (OIXIV) |
AR (Shaker et al., 2015). |
TMEM38B (Shaker et al., 2015). |
9q31.2(OMIM* 611236). |
Unknown |
Sever fractures, osteopenia, normal teeth, sclerae of eye and hearing(Shaheen et al., 2012). |
|
22 |
Osteogenesis imperfecta type XV (OIXV) |
AR/AD (Shaker et al., 2015). |
WNT1 (Shaker et al., 2015). |
12q13.12 (OMIM# 615220). |
Unknown |
Recurrent fractures, bone deformity, significant reduction of bone density, short stature, development issues and brain anomalies and normal hearing (Pyott et al., 2013). |
|
23 |
Hereditary multiple exostosis (HME) |
AD (Alvarez et al., 2007). |
EXT1 and EXT2 (Alvarez et al., 2007). |
8q24 (EXT1), 11p11-13 (EXT2) (Francannet et al., 2001). |
1/100,000 (Pannier and Legeai-Mallet., 2008). |
Shortening of stature, limb problem, inconsistencies, valgus abnormalities of knee and ankle, irregularity of the pelvic, pectoral, bending of radius, eccentricity ulnar, subluxation of the radiocapitellar joint (Stieber and Dormans, 2005). |
|
24 |
Frontometaphyseal dysplasia (FMD) |
X-linked dominant (Morava et al., 2003). |
FLNA (Robertson et al., 2006). |
Xq28 (Stephen et al., 2003). |
More than 100 cases (Morava et al., 2003). |
Thickening of bone tissue of skull and molding abnormalities of the tubular bones, trachea -bronchial, cardiac, and urology abnormalities (Robertson et al., 2006), skeleton and connective tissue problems significant supr-aorbital edges, cranial hyperostosis, flared metaphyses (Morava et al., 2003). |
|
25 |
Ellis-van creveld syndrome (EVC)/ Weyers acrofacial dysostosis (WAD) |
AR (Ruiz-Perez and Goodship, 2009). |
EVC and EVC2 (Hills et al., 2011). |
4p16 (Baujat and Le Merrer, 2007). |
Unknown (Baujat and Le Merrer, 2007). |
Bone dysplasia (stippling of epiphyses), polydactyl, abnormal nail, acrofacial anomalies cardio-vascular malformations(Ruiz-Perez and Goodship, 2009), narrow thorax, shortness of long bones, hexadactyly, cardiac flaws, short stature and ribs, heart defects, especially anomalies of atrial septation (Baujat and Le Merrer, 2007). |
|
26 |
Ghosal hematodiaphseal dysplasia (GHDD) |
AD (Isidor et al., 2007). |
TBXAS1 (Genevieve, Proulle, et al., 2008). |
7q33-34 (Isidor et al., 2007). |
39 cases (Mondal et al., 2007). |
Severe anemia, leukopenia, wide medullary cavities with distinct cortical hyperosthosis, bone marrow hypocellular thick bone anomalies (Gumruket al., 1993), increased bone density(Genevieve et al., 2008). |
|
27 |
Greenberg dysplasia (GRBGD) |
AR (Waterham et al., 2003). |
LBR (Clayton et al., 2010). |
1q41-43 (Hoffmann et al., 2002). |
Unknown |
Abnormally chondro-osseous short limbs, accumulation of calcium, long bones shorts, disorders of pelvis, ribs, trachea, trachea (Trajkovski et al., 2002). |
|
28 |
Langer mesomelic dysplasia (LMD) |
AR (OMIM # 249700). |
SHOX (Aggarwal, and Venkat, 2014). |
Xp22.33 (Thomas et al., 2004). |
Unknown |
Short ulnae, bent and synostoses of tibia, fibula, and short, anomalies of cuboid bone of feet and talus(Kantaputra et al., 1992), skeleton abnormalities (Aggarwal et al., 2014). |
|
29 |
Fibrodysplasia ossificans progressiva (FOP) |
AD (Feldman et al., 2000). |
ACVR1/ALK2 (Kaplan et al., 2008). |
4q27-31 (Feldman et al., 2000). |
1/2,000,000 (Pignolo et al., 2011). |
Unadorned disabling, muscles ossification, large tendon, ligament and fascia, toes extra-skeleton bone formation, fibro-proliferative on the upper and back neck (Feldman et al., 2000). |
|
30 |
Juvenile paget's disease (JPD) |
AR (Whyte et al., 2002). |
TNFRSF11B (Saki et al., 2013). |
8q24 (Cundy et al., 2002). |
51 cases (Indumathi et al., 2009). |
Pain in bones and fractures abnormalities and weakness of large and thick bones (Saki et al., 2013). |



